Synapse and AD Knowledge Portal
Overview
Teaching: 45 min
Exercises: 15 minQuestions
How to work with Synapse R client?
How to work with data in AD Knowledge Portal?
Objectives
Explain how to use Synapser Package.
Demonstrate how to locate data and metadata in the Portal.
Demonstrate how to download data from the Portal programmatically.
Author: Sage Bionetworks
Setup
Install and load packages
If you haven’t already, install synapser (the Synapse R client), as well as the tidyverse family of packages.
# install synapser
install.packages("synapser", repos = c("http://ran.synapse.org", "http://cran.fhcrc.org"))
# install tidyverse if you don't already have it
install.packages("tidyverse")
We will also use the BioconductoR package manager to install biomaRt, which will help us with gene count data later.
#install.packages("XML")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("biomaRt")
Load libraries
library(synapser)
library(tidyverse)
library(biomaRt)
Login to Synapse
Next, you will need to log in to your Synapse account.
Login option 1: Synapser takes credentials from your Synapse web session
If you are logged into the Synapse web browser, synapser will automatically use your login credentials to log you in during your R session! All you have to do is:
synLogin()
If for whatever reason that didn’t work, try one of these options:
Login option 2: Synapse username and password
In the code below, replace the <> with your Synapse username and password.
synLogin("<username>", "<password>")Login option 3: Synapse PAT
If you usually log in to Synapse with your Google account, you will need to use a Synapser Personal Access Token (PAT) to log in with the R client. Follow these instructions to generate a personal access token, then paste the PAT into the code below. Make sure you scope your access token to allow you to View, Download, and Modify.
synLogin(authToken = "<paste your personal access token here>")For more information on managing Synapse credentials with
synapser, see the documentation here.
Download data
While you can always download data from the AD Portal website via your web browser, it’s usually faster and often more convenient to download data programmatically.
Download a single file
To download a single file from the AD Knowledge Portal, you can click the linked file name to go to a page in the Synapse platform where that file is stored. Using the synID on that page, you can call the synGet() function from synapser to download the file.
Exercise 1: Use Explore Data to find processed RNAseq data from the Jax.IU.Pitt_5XFAD Study
This filters the table to a single file. In the “Id” column for this htseqcounts_5XFAD.txt file, there is a unique Synapse ID (synID).
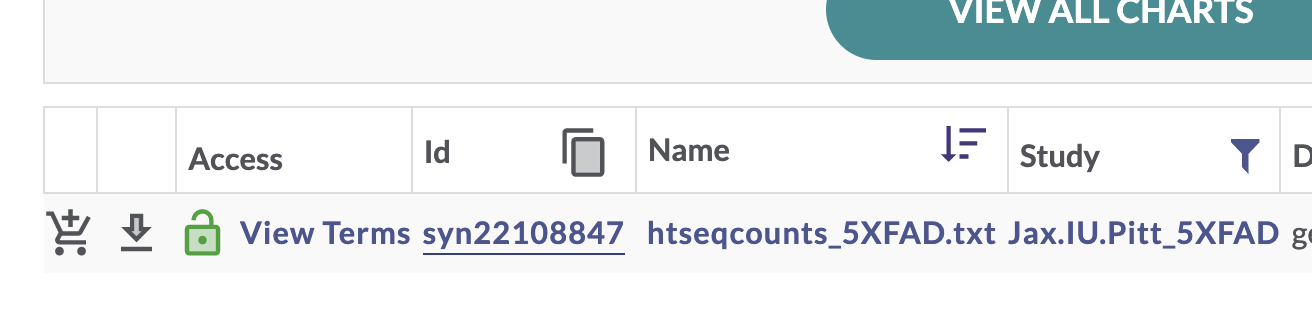
We can then use that synID to download the file.
counts_id <- "syn22108847"
synGet(counts_id, downloadLocation = "../data/")
Bulk download files
Exercise 2: Use Explore Studies to find all metadata files from the Jax.IU.Pitt_5XFAD study
Use the facets and search bar to look for data you want to download from the AD Knowledge Portal. Once you’ve identified the files you want, click on the download arrow icon on the top right of the Explore Data table and select “Programmatic Options” from the drop-down menu.
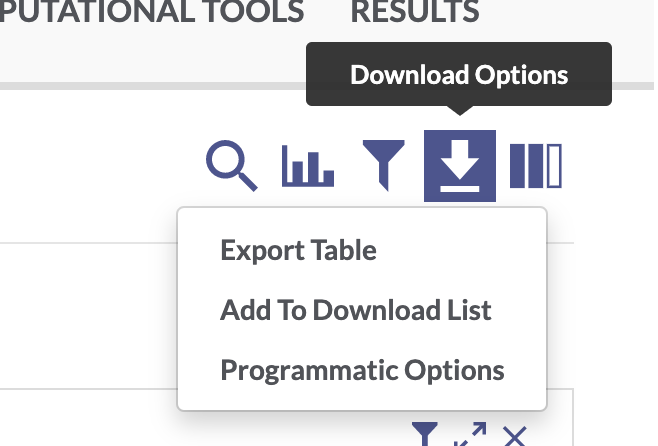
In the window that pops up, select the “R” tab from the top menu bar. This will display some R code that constructs a SQL query of the Synapse data table that drives the AD Knowledge Portal. This query will allow us to download only the files that meet our search criteria.
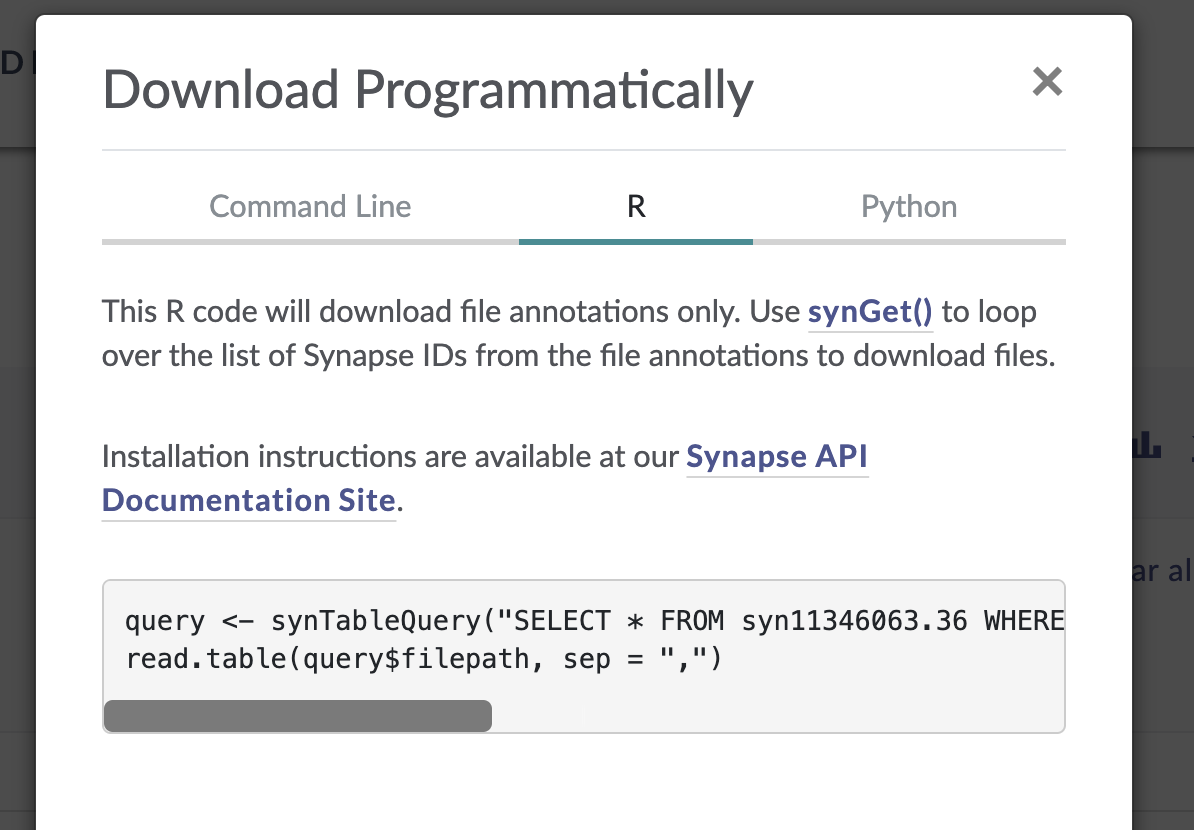
The function synTableQuery() returns a Synapse object wrapper around a CSV file that is automatically downloaded to a Synapse cache directory .synapseCache in your home directory. You can use query$filepath to see the path to the file in the Synapse cache.
# download the results of the filtered table query
query <- synTableQuery("SELECT * FROM syn11346063.37 WHERE ( ( `study` HAS ( 'Jax.IU.Pitt_5XFAD' ) ) AND ( `resourceType` = 'metadata' ) )")
# view the file path of the resulting csv
query$filepath
[1] "/Users/auyar/.synapseCache/628/125732628/SYNAPSE_TABLE_QUERY_125732628.csv"
We’ll use read.csv to read the CSV file into R (although the provided read.table or any other base R version is also fine!). We can explore the download_table object and see that it contains information on all of the AD Portal data files we want to download. Some columns like the “id” and “parentId” columns contain info about where the file is in Synapse, and some columns contain AD Portal annotations for each file, like “dataType”, “specimenID”, and “assay”. This annotation table will later allow us to link downloaded files to additional metadata variables!
# read in the table query csv file
download_table <- read_csv(query$filepath, show_col_types = FALSE)
download_table
# A tibble: 5 × 45
ROW_ID ROW_VERSION ROW_ETAG id name study dataType assay organ tissue
<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <chr> <lgl> <lgl>
1 22094731 2 84e4bc38-9… syn2… Jax.… "[\"… "[\"ima… "[\"… NA NA
2 22094732 2 266ec572-1… syn2… Jax.… "[\"… "[\"ima… "[\"… NA NA
3 22103212 3 7229ee91-4… syn2… Jax.… "[\"… <NA> <NA> NA NA
4 22103213 3 ce970c4c-d… syn2… Jax.… "[\"… <NA> <NA> NA NA
5 22110328 4 20cf3097-4… syn2… Jax.… "[\"… "[\"gen… "[\"… NA NA
# ℹ 35 more variables: species <chr>, sex <lgl>, consortium <chr>, grant <chr>,
# modelSystemName <lgl>, treatmentType <lgl>, specimenID <lgl>,
# individualID <lgl>, individualIdSource <lgl>, specimenIdSource <lgl>,
# resourceType <chr>, dataSubtype <lgl>, metadataType <chr>,
# assayTarget <lgl>, analysisType <lgl>, cellType <lgl>,
# nucleicAcidSource <lgl>, fileFormat <chr>, group <lgl>,
# isModelSystem <lgl>, isConsortiumAnalysis <lgl>, isMultiSpecimen <lgl>, …
Finally, we use a mapping function from the purrr package to loop through the “id” column and apply the synGet() function to each file’s synID. In this case, we use purrr::walk() because it lets us call synGet() for its side effect (downloading files to a location we specify), and returns nothing.
# loop through the column of synIDs and download each file
purrr::walk(download_table$id, ~synGet(.x, downloadLocation = "../data/"))
Congratulations, you have bulk downloaded files from the AD Knowledge Portal!
An important note: for situations where you are downloading many large files, the R client performs substantially slower than the command line client or the Python client. In these cases, you can use the instructions and code snippets for the command line or Python client provided in the “Programmatic Options” menu.
Working with AD Portal metadata
Metadata basics
We have now downloaded several metadata files and an RNAseq counts file from the portal. For our next exercises, we want to read those files in as R data so we can work with them.
We can see from the download_table we got during the bulk download step that we have five metadata files. Two of these should be the individual and biospecimen files, and three of them are assay metadata files.
#
download_table %>%
dplyr::select(name, metadataType, assay)
# A tibble: 5 × 3
name metadataType assay
<chr> <chr> <chr>
1 Jax.IU.Pitt_5XFAD_assay_autorad_metadata.csv assay "[\"autoradiography…
2 Jax.IU.Pitt_5XFAD_assay_PET_metadata.csv assay "[\"Positron Emissi…
3 Jax.IU.Pitt_5XFAD_individual_metadata.csv individual <NA>
4 Jax.IU.Pitt_5XFAD_biospecimen_metadata.csv biospecimen <NA>
5 Jax.IU.Pitt_5XFAD_assay_RNAseq_metadata.csv assay "[\"rnaSeq\"]"
We are only interested in RNAseq data, so we will only read in the individual, biospecimen, and RNAseq assay metadata files.
# counts matrix
counts <- read_tsv("../data/htseqcounts_5XFAD.txt", show_col_types = FALSE)
# individual metadata
ind_meta <- read_csv("../data/Jax.IU.Pitt_5XFAD_individual_metadata.csv", show_col_types = FALSE)
# biospecimen metadata
bio_meta <- read_csv("../data/Jax.IU.Pitt_5XFAD_biospecimen_metadata.csv", show_col_types = FALSE)
#assay metadata
rna_meta <- read_csv("../data/Jax.IU.Pitt_5XFAD_assay_RNAseq_metadata.csv", show_col_types = FALSE)
Let’s examine the data and metadata files a bit before we begin our analyses.
Counts data
# Calling a tibble object will print the first ten rows in a nice tidy output; doing the same for a base R dataframe will print the whole thing until it runs out of memory. If you want to inspect a large dataframe, use `head(df)`
counts
# A tibble: 55,489 × 73
gene_id `32043rh` `32044rh` `32046rh` `32047rh` `32048rh` `32049rh` `32050rh`
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 ENSG00… 22554 0 0 0 16700 0 0
2 ENSG00… 344489 4 0 1 260935 6 8
3 ENSMUS… 5061 3483 3941 3088 2756 3067 2711
4 ENSMUS… 0 0 0 0 0 0 0
5 ENSMUS… 208 162 138 127 95 154 165
6 ENSMUS… 44 17 14 28 23 24 14
7 ENSMUS… 143 88 121 117 115 109 75
8 ENSMUS… 22 6 10 11 11 19 24
9 ENSMUS… 7165 5013 5581 4011 4104 5254 4345
10 ENSMUS… 3728 2316 2238 1965 1822 1999 1809
# ℹ 55,479 more rows
# ℹ 65 more variables: `32052rh` <dbl>, `32053rh` <dbl>, `32057rh` <dbl>,
# `32059rh` <dbl>, `32061rh` <dbl>, `32062rh` <dbl>, `32065rh` <dbl>,
# `32067rh` <dbl>, `32068rh` <dbl>, `32070rh` <dbl>, `32073rh` <dbl>,
# `32074rh` <dbl>, `32075rh` <dbl>, `32078rh` <dbl>, `32081rh` <dbl>,
# `32088rh` <dbl>, `32640rh` <dbl>, `46105rh` <dbl>, `46106rh` <dbl>,
# `46107rh` <dbl>, `46108rh` <dbl>, `46109rh` <dbl>, `46110rh` <dbl>, …
The data file has a column of ENSEMBL gene ids and then a bunch of columns with count data, where the column headers correspond to the specimenIDs. These specimenIDs should all be in the RNAseq assay metadata file, so let’s check.
# what does the RNAseq assay metadata look like?
rna_meta
# A tibble: 72 × 12
specimenID platform RIN rnaBatch libraryBatch sequencingBatch libraryPrep
<chr> <chr> <lgl> <dbl> <dbl> <dbl> <chr>
1 32043rh IlluminaN… NA 1 1 1 polyAselec…
2 32044rh IlluminaN… NA 1 1 1 polyAselec…
3 32046rh IlluminaN… NA 1 1 1 polyAselec…
4 32047rh IlluminaN… NA 1 1 1 polyAselec…
5 32049rh IlluminaN… NA 1 1 1 polyAselec…
6 32057rh IlluminaN… NA 1 1 1 polyAselec…
7 32061rh IlluminaN… NA 1 1 1 polyAselec…
8 32065rh IlluminaN… NA 1 1 1 polyAselec…
9 32067rh IlluminaN… NA 1 1 1 polyAselec…
10 32070rh IlluminaN… NA 1 1 1 polyAselec…
# ℹ 62 more rows
# ℹ 5 more variables: libraryPreparationMethod <lgl>, isStranded <lgl>,
# readStrandOrigin <lgl>, runType <chr>, readLength <dbl>
# are all the column headers from the counts matrix (except the first "gene_id" column) in the assay metadata?
all(colnames(counts[-1]) %in% rna_meta$specimenID)
[1] TRUE
Assay metadata
The assay metadata contains information about how data was generated on each sample in the assay. Each specimenID represents a unique sample. We can use some tools from dplyr to explore the metadata.
# how many unique specimens were sequenced?
n_distinct(rna_meta$specimenID)
[1] 72
# were the samples all sequenced on the same platform?
distinct(rna_meta, platform)
# A tibble: 1 × 1
platform
<chr>
1 IlluminaNovaseq6000
# were there multiple sequencing batches reported?
distinct(rna_meta, sequencingBatch)
# A tibble: 1 × 1
sequencingBatch
<dbl>
1 1
Biospecimen metadata
The biospecimen metadata contains specimen-level information, including organ and tissue the specimen was taken from, how it was prepared, etc. Each specimenID is mapped to an individualID.
# all specimens from the RNAseq assay metadata file should be in the biospecimen file
all(rna_meta$specimenID %in% bio_meta$specimenID)
[1] TRUE
# but the biospecimen file also contains specimens from different assays
all(bio_meta$specimenID %in% rna_meta$specimenID)
[1] FALSE
Individual metadata
The individual metadata contains information about all the individuals in the study, represented by unique individualIDs. For humans, this includes information on age, sex, race, diagnosis, etc. For MODEL-AD mouse models, the individual metadata has information on model genotypes, stock numbers, diet, and more.
# all individualIDs in the biospecimen file should be in the individual file
all(bio_meta$individualID %in% ind_meta$individualID)
[1] FALSE
# which model genotypes are in this study?
distinct(ind_meta, genotype)
# A tibble: 3 × 1
genotype
<chr>
1 <NA>
2 5XFAD_carrier
3 5XFAD_noncarrier
Joining metadata
We use the three-file structure for our metadata because it allows us to store metadata for each study in a tidy format. Every line in the assay and biospecimen files represents a unique specimen, and every line in the individual file represents a unique individual. This means the files can be easily joined by specimenID and individualID to get all levels of metadata that apply to a particular data file. We will use the left_join() function from the dplyr package, and the %>% operator from the magrittr package. If you are unfamiliar with the pipe, think of it as a shorthand for “take this (the preceding object) and do that (the subsequent command)”. See here for more info on piping in R.
# join all the rows in the assay metadata that have a match in the biospecimen metadata
joined_meta <- rna_meta %>% #start with the rnaseq assay metadata
left_join(bio_meta, by = "specimenID") %>% #join rows from biospecimen that match specimenID
left_join(ind_meta, by = "individualID") # join rows from individual that match individualID
joined_meta
# A tibble: 144 × 50
specimenID platform RIN rnaBatch libraryBatch sequencingBatch libraryPrep
<chr> <chr> <lgl> <dbl> <dbl> <dbl> <chr>
1 32043rh IlluminaN… NA 1 1 1 polyAselec…
2 32043rh IlluminaN… NA 1 1 1 polyAselec…
3 32044rh IlluminaN… NA 1 1 1 polyAselec…
4 32044rh IlluminaN… NA 1 1 1 polyAselec…
5 32046rh IlluminaN… NA 1 1 1 polyAselec…
6 32046rh IlluminaN… NA 1 1 1 polyAselec…
7 32047rh IlluminaN… NA 1 1 1 polyAselec…
8 32047rh IlluminaN… NA 1 1 1 polyAselec…
9 32049rh IlluminaN… NA 1 1 1 polyAselec…
10 32049rh IlluminaN… NA 1 1 1 polyAselec…
# ℹ 134 more rows
# ℹ 43 more variables: libraryPreparationMethod <lgl>, isStranded <lgl>,
# readStrandOrigin <lgl>, runType <chr>, readLength <dbl>,
# individualID <dbl>, specimenIdSource <chr>, organ <chr>, tissue <chr>,
# BrodmannArea <lgl>, sampleStatus <chr>, tissueWeight <lgl>,
# tissueVolume <lgl>, nucleicAcidSource <lgl>, cellType <lgl>,
# fastingState <lgl>, isPostMortem <lgl>, samplingAge <lgl>, ...1 <dbl>, …
We now have a very wide dataframe that contains all the available metadata on each specimen in the RNAseq data from this study. This procedure can be used to join the three types of metadata files for every study in the AD Knowledge Portal, allowing you to filter individuals and specimens as needed based on your analysis criteria!
# convert columns of strings to month-date-year format
joined_meta_time <- joined_meta %>%
# convert numeric ages to timepoint categories
mutate(timepoint = case_when(ageDeath > 10 ~ "12 mo",
ageDeath < 10 & ageDeath > 5 ~ "6 mo",
ageDeath < 5 ~ "4 mo"))
covars_5XFAD <- joined_meta_time %>%
dplyr::select(individualID, specimenID, sex, genotype, timepoint) %>% distinct() %>% as.data.frame()
rownames(covars_5XFAD) <- covars_5XFAD$specimenID
We will save joined_meta for the next lesson.
saveRDS(covars_5XFAD, file = "../data/covars_5XFAD.rds")
Single-specimen files
For files that contain data from a single specimen (e.g. raw sequencing files, raw mass spectra, etc.), we can use the Synapse annotations to associate these files with the appropriate metadata.
Excercise 3: Use Explore Data to find all RNAseq files from the Jax.IU.Pitt_5XFAD study.
If we filter for data where Study = “Jax.IU.Pitt_5XFAD” and Assay = “rnaSeq” we will get a list of 148 files, including raw fastqs and processed counts data.
Synapse entity annotations
We can use the function synGetAnnotations to view the annotations associated with any file without downloading the file.
# the synID of a random fastq file from this list
random_fastq <- "syn22108503"
# extract the annotations as a nested list
fastq_annotations <- synGetAnnotations(random_fastq)
fastq_annotations
$sex
[1] "female"
$room
[1] "JAX_MGL373"
$assay
[1] "rnaSeq"
$grant
[1] "U54AG054345"
$organ
[1] "brain"
$study
[1] "Jax.IU.Pitt_5XFAD"
$tissue
[1] "right cerebral hemisphere"
$bedding
[1] "aspen"
$birthID
[1] "RMO1223"
$climbID
[1] "298456"
$species
[1] "Mouse"
$waterpH
[1] 2.85
$ageDeath
[1] 10.81967
$dataType
[1] "geneExpression"
$genotype
[1] "5XFAD_carrier"
$matingID
[1] "M-108-17"
$dateBirth
[1] "1521417600000"
$consortium
[1] "MODEL-AD"
$fileFormat
[1] "fastq"
$generation
[1] "N1F3"
$rodentDiet
[1] "0.06"
$specimenID
[1] "32043rh"
$brainWeight
[1] 0.503
$dataSubtype
[1] "raw"
$microchipID
[1] "288646853"
$stockNumber
[1] "8730"
$individualID
[1] "32043"
$officialName
[1] "B6.Cg-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax"
$resourceType
[1] "experimentalData"
$rodentWeight
[1] 28.76
$ageDeathUnits
[1] "months"
$isModelSystem
[1] FALSE
$materialOrigin
[1] "JAX"
$isMultiSpecimen
[1] FALSE
$modelSystemName
[1] "5XFAD"
$modelSystemType
[1] "animal"
$nucleicAcidSource
[1] "bulk cell"
$genotypeBackground
[1] "C57BL6J"
$individualIdSource
[1] "JAX"
$individualCommonGenotype
[1] "5XFAD"
The file annotations let us see which study the file is associated with (Jax.IU.Pitt.5XFAD), which species it’s from (Mouse), which assay generated the file (rnaSeq), and a whole bunch of other properties. Most importantly, single-specimen files are annotated with with the specimenID of the specimen in the file, and the individualID of the individual that specimen was taken from. We can use these annotations to link files to the rest of the metadata, including metadata that is not in annotations. This is especially helpful for human studies, as potentially identifying information like age, race, and diagnosis is not included in file annotations.
# find records belonging to the individual this file maps to in our joined metadata
joined_meta %>%
filter(individualID == fastq_annotations$individualID[[1]])
# A tibble: 2 × 50
specimenID platform RIN rnaBatch libraryBatch sequencingBatch libraryPrep
<chr> <chr> <lgl> <dbl> <dbl> <dbl> <chr>
1 32043rh IlluminaNo… NA 1 1 1 polyAselec…
2 32043rh IlluminaNo… NA 1 1 1 polyAselec…
# ℹ 43 more variables: libraryPreparationMethod <lgl>, isStranded <lgl>,
# readStrandOrigin <lgl>, runType <chr>, readLength <dbl>,
# individualID <dbl>, specimenIdSource <chr>, organ <chr>, tissue <chr>,
# BrodmannArea <lgl>, sampleStatus <chr>, tissueWeight <lgl>,
# tissueVolume <lgl>, nucleicAcidSource <lgl>, cellType <lgl>,
# fastingState <lgl>, isPostMortem <lgl>, samplingAge <lgl>, ...1 <dbl>,
# climbID <dbl>, microchipID <dbl>, birthID <chr>, matingID <chr>, …
Annotations during bulk download
When bulk downloading many files, the best practice is to preserve the download manifest that is generated which lists all the files, their synIDs, and all their annotations. If using the Synapse R client, follow the instructions in the Bulk download files section above.
If we use the “Programmatic Options” tab in the AD Portal download menu to download all 148 rnaSeq files from the 5XFAD study, we would get a table query that looks like this:
query <- synTableQuery("SELECT * FROM syn11346063.37 WHERE ( ( \"study\" HAS ( 'Jax.IU.Pitt_5XFAD' ) ) AND ( \"assay\" HAS ( 'rnaSeq' ) ) )")
As we saw previously, this downloads a csv file with the results of our AD Portal query. Opening that file lets us see which specimens are associated with which files:
#
annotations_table <- read_csv(query$filepath, show_col_types = FALSE)
annotations_table
# A tibble: 148 × 45
ROW_ID ROW_VERSION ROW_ETAG id name study dataType assay organ tissue
<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
1 22108503 1 458ff182-… syn2… 3204… "[\"… "[\"gen… "[\"… brain "[\"r…
2 22108508 1 163d5067-… syn2… 3204… "[\"… "[\"gen… "[\"… brain "[\"r…
3 22108512 1 d02e16c5-… syn2… 3204… "[\"… "[\"gen… "[\"… brain "[\"r…
4 22108519 1 59eba082-… syn2… 3204… "[\"… "[\"gen… "[\"… brain "[\"r…
5 22108525 1 9fed0677-… syn2… 3204… "[\"… "[\"gen… "[\"… brain "[\"r…
6 22108530 1 47cff701-… syn2… 3205… "[\"… "[\"gen… "[\"… brain "[\"r…
7 22108534 1 03af7c0a-… syn2… 3206… "[\"… "[\"gen… "[\"… brain "[\"r…
8 22108539 1 502d1468-… syn2… 3206… "[\"… "[\"gen… "[\"… brain "[\"r…
9 22108543 1 47b8ffe9-… syn2… 3206… "[\"… "[\"gen… "[\"… brain "[\"r…
10 22108550 1 471ec9a1-… syn2… 3207… "[\"… "[\"gen… "[\"… brain "[\"r…
# ℹ 138 more rows
# ℹ 35 more variables: species <chr>, sex <chr>, consortium <chr>, grant <chr>,
# modelSystemName <chr>, treatmentType <lgl>, specimenID <chr>,
# individualID <dbl>, individualIdSource <chr>, specimenIdSource <lgl>,
# resourceType <chr>, dataSubtype <chr>, metadataType <chr>,
# assayTarget <lgl>, analysisType <lgl>, cellType <lgl>,
# nucleicAcidSource <chr>, fileFormat <chr>, group <lgl>, …
You could then use purrr::walk(download_table$id, ~synGet(.x, downloadLocation = <your-download-directory>)) to walk through the column of synIDs and download all 148 files. However, because these are large files, it might be preferable to use the Python client or command line client for increased speed.
Once you’ve downloaded all the files in the id column, you can link those files to their annotations by the name column.
# We'll use the "random fastq" that we got annotations for earlier
# To avoid downloading the whole 3GB file, we'll use synGet with "downloadFile = FALSE" to get only the Synapse entity information, rather than the file.
# If we downloaded the actual file, we could find it in the directory and search using the filename. Since we're just downloading the Synapse entity wrapper object, we'll use the file name listed in the object properties.
fastq <- synGet(random_fastq, downloadFile = FALSE)
# filter the annotations table to rows that match the fastq filename
annotations_table %>%
filter(name == fastq$properties$name)
# A tibble: 1 × 45
ROW_ID ROW_VERSION ROW_ETAG id name study dataType assay organ tissue
<dbl> <dbl> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
1 22108503 1 458ff182-d… syn2… 3204… "[\"… "[\"gen… "[\"… brain "[\"r…
# ℹ 35 more variables: species <chr>, sex <chr>, consortium <chr>, grant <chr>,
# modelSystemName <chr>, treatmentType <lgl>, specimenID <chr>,
# individualID <dbl>, individualIdSource <chr>, specimenIdSource <lgl>,
# resourceType <chr>, dataSubtype <chr>, metadataType <chr>,
# assayTarget <lgl>, analysisType <lgl>, cellType <lgl>,
# nucleicAcidSource <chr>, fileFormat <chr>, group <lgl>,
# isModelSystem <lgl>, isConsortiumAnalysis <lgl>, isMultiSpecimen <lgl>, …
Multispecimen files
Multispecimen files in the AD Knowledge Portal are files that contain data or information from more than one specimen. They are not annotated with individualIDs or specimenIDs, since these files may contain numbers of specimens that exceed the annotation limits. These files are usually processed or summary data (gene counts, peptide quantifications, etc), and are always annotated with isMultiSpecimen = TRUE.
If we look at the processed data files in the table of 5XFAD RNAseq file annotations we just downloaded, we will see that it isMultiSpecimen = TRUE, but individualID and specimenID are blank:
#
annotations_table %>%
filter(fileFormat == "txt") %>%
dplyr::select(name, individualID, specimenID, isMultiSpecimen)
# A tibble: 3 × 4
name individualID specimenID isMultiSpecimen
<chr> <dbl> <chr> <lgl>
1 htseqcounts_5XFAD.txt NA <NA> TRUE
2 tpm_gene_5XFAD.txt NA <NA> TRUE
3 tpm_isoform_5XFAD.txt NA <NA> TRUE
The multispecimen file should contain a row or column of specimenIDs that correspond to the specimenIDs used in a study’s metadata, as we have seen with the 5XFAD counts file.
# In this example, we take a slice of the counts data to reduce computation, transpose it so that each row represents a single specimen, and then join it to the joined metadata by the specimenID
counts %>%
slice_head(n = 5) %>%
t() %>%
as_tibble(rownames = "specimenID") %>%
left_join(joined_meta, by = "specimenID")
Warning: The `x` argument of `as_tibble.matrix()` must have unique column names if `.name_repair` is
omitted as of tibble 2.0.0.
ℹ Using compatibility `.name_repair`.
This warning is displayed once every 8 hours.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was generated.
# A tibble: 145 × 55
specimenID V1 V2 V3 V4 V5 platform RIN rnaBatch libraryBatch
<chr> <chr> <chr> <chr> <chr> <chr> <chr> <lgl> <dbl> <dbl>
1 gene_id "ENS… "ENS… "ENS… "ENS… "ENS… <NA> NA NA NA
2 32043rh " 22… "344… " 5… " … " … Illumin… NA 1 1
3 32043rh " 22… "344… " 5… " … " … Illumin… NA 1 1
4 32044rh " … " … "348… " … " 16… Illumin… NA 1 1
5 32044rh " … " … "348… " … " 16… Illumin… NA 1 1
6 32046rh " … " … "394… " … " 13… Illumin… NA 1 1
7 32046rh " … " … "394… " … " 13… Illumin… NA 1 1
8 32047rh " … " … "308… " … " 12… Illumin… NA 1 1
9 32047rh " … " … "308… " … " 12… Illumin… NA 1 1
10 32048rh " 16… "260… " 2… " … " … Illumin… NA 1 1
# ℹ 135 more rows
# ℹ 45 more variables: sequencingBatch <dbl>, libraryPrep <chr>,
# libraryPreparationMethod <lgl>, isStranded <lgl>, readStrandOrigin <lgl>,
# runType <chr>, readLength <dbl>, individualID <dbl>,
# specimenIdSource <chr>, organ <chr>, tissue <chr>, BrodmannArea <lgl>,
# sampleStatus <chr>, tissueWeight <lgl>, tissueVolume <lgl>,
# nucleicAcidSource <lgl>, cellType <lgl>, fastingState <lgl>, …
Challenge 1
Download the raw read counts and the metadata from the Jax.IU.Pitt_APOE4.TREM2.R47H study (syn22107627, syn23613784).
Solution to Challenge 1
counts_id <- "syn22107627" synGet(counts_id, downloadLocation = "../data/") metadata_id <- 'syn23613784' synGet(metadata_id, downloadLocation = "../data/")
Key Points
Use your Synapse login credentials to access the Portal.
Use Synapser package to download data from the Portal.