Differential Expression Analysis
Overview
Teaching: 40 min
Exercises: 10 minQuestions
What transcriptomic changes we observe in mouse models carrying AD-related mutations?
Objectives
Read in a count matrix and metadata.
Understand the data from AD mouse models
Format the data for differential analysis
Perform differential analysis using DESeq2.
Pathway enrichment of differentially expressed genes
Save data for next lessons
Author: Ravi Pandey, Jackson Laboratory
LOAD libraries
suppressPackageStartupMessages(library("DESeq2"))
suppressPackageStartupMessages(library("ggplot2"))
suppressPackageStartupMessages(library("AnnotationDbi"))
suppressPackageStartupMessages(library("org.Mm.eg.db"))
suppressPackageStartupMessages(library("GO.db"))
suppressPackageStartupMessages(library("EnhancedVolcano"))
suppressPackageStartupMessages(library(tidyverse))
suppressPackageStartupMessages(library(dplyr))
suppressPackageStartupMessages(library(clusterProfiler))
Reading Gene Expression Count matrix from Previous Lesson
In this lesson, we will use the raw counts matrix and metadata downloaded in the previous lesson and will perform differential expression analysis.
RNA-Seq data from 5xFAD mouse models
counts <- read.delim("../data/htseqcounts_5XFAD.txt", check.names = FALSE)
Reading Sample Metadata from Previous Lesson
covars <- readRDS("../data/covars_5XFAD.rds")
Let’s explore the data:
Let’s look at the top of the metadata.
head(covars)
individualID specimenID sex genotype timepoint
32043rh 32043 32043rh female 5XFAD_carrier 12 mo
32044rh 32044 32044rh male 5XFAD_noncarrier 12 mo
32046rh 32046 32046rh male 5XFAD_noncarrier 12 mo
32047rh 32047 32047rh male 5XFAD_noncarrier 12 mo
32049rh 32049 32049rh female 5XFAD_noncarrier 12 mo
32057rh 32057 32057rh female 5XFAD_noncarrier 12 mo
identify distinct groups using sample metadata
distinct(covars, sex, genotype, timepoint)
sex genotype timepoint
32043rh female 5XFAD_carrier 12 mo
32044rh male 5XFAD_noncarrier 12 mo
32049rh female 5XFAD_noncarrier 12 mo
46105rh female 5XFAD_noncarrier 6 mo
46108rh male 5XFAD_noncarrier 6 mo
46131rh female 5XFAD_noncarrier 4 mo
46877rh male 5XFAD_noncarrier 4 mo
46887rh female 5XFAD_carrier 4 mo
32053rh male 5XFAD_carrier 12 mo
46111rh female 5XFAD_carrier 6 mo
46865rh male 5XFAD_carrier 6 mo
46866rh male 5XFAD_carrier 4 mo
We’re going to explore the data further using a series of Challenges. You will be asked to look at the contents of some of the columns to see how the data are distributed.
Challenge 1
How many mice were used to produce this data?
Solution to Challenge 1
covars %>% group_by(sex,genotype,timepoint) %>% count() dplyr::count(metadata, sex, genotype,timepoint)
How many rows and columns are there in counts?
dim(counts)
[1] 55489 73
In the counts matrix, genes are in rows and samples are in columns. Let’s look at the first few rows.
head(counts,n=5)
gene_id 32043rh 32044rh 32046rh 32047rh 32048rh 32049rh 32050rh
1 ENSG00000080815 22554 0 0 0 16700 0 0
2 ENSG00000142192 344489 4 0 1 260935 6 8
3 ENSMUSG00000000001 5061 3483 3941 3088 2756 3067 2711
4 ENSMUSG00000000003 0 0 0 0 0 0 0
5 ENSMUSG00000000028 208 162 138 127 95 154 165
32052rh 32053rh 32057rh 32059rh 32061rh 32062rh 32065rh 32067rh 32068rh
1 19748 14023 0 17062 0 15986 10 0 18584
2 337456 206851 1 264748 0 252248 172 4 300398
3 3334 3841 4068 3306 4076 3732 3940 4238 3257
4 0 0 0 0 0 0 0 0 0
5 124 103 164 116 108 134 204 239 148
32070rh 32073rh 32074rh 32075rh 32078rh 32081rh 32088rh 32640rh 46105rh
1 1 0 0 22783 17029 16626 15573 12721 4
2 4 2 9 342655 280968 258597 243373 188818 19
3 3351 3449 4654 4844 3132 3334 3639 3355 4191
4 0 0 0 0 0 0 0 0 0
5 159 167 157 211 162 149 160 103 158
46106rh 46107rh 46108rh 46109rh 46110rh 46111rh 46112rh 46113rh 46115rh
1 0 0 0 0 0 17931 0 19087 0
2 0 0 1 5 1 293409 8 273704 1
3 3058 4265 3248 3638 3747 3971 3192 3805 3753
4 0 0 0 0 0 0 0 0 0
5 167 199 113 168 175 203 158 108 110
46121rh 46131rh 46132rh 46134rh 46138rh 46141rh 46142rh 46862rh 46863rh
1 0 0 12703 18833 0 18702 17666 0 14834
2 0 1 187975 285048 0 284499 250600 0 218494
3 4134 3059 3116 3853 3682 2844 3466 3442 3300
4 0 0 0 0 0 0 0 0 0
5 179 137 145 183 171 138 88 154 157
46865rh 46866rh 46867rh 46868rh 46871rh 46872rh 46873rh 46874rh 46875rh
1 10546 10830 10316 10638 15248 0 0 11608 11561
2 169516 152769 151732 190150 229063 6 1 165941 171303
3 3242 3872 3656 3739 3473 3154 5510 3657 4121
4 0 0 0 0 0 0 0 0 0
5 131 152 152 155 140 80 240 148 112
46876rh 46877rh 46878rh 46879rh 46881rh 46882rh 46883rh 46884rh 46885rh
1 0 0 12683 15613 0 14084 20753 0 0
2 0 2 183058 216122 0 199448 306081 0 5
3 3422 3829 3996 4324 2592 2606 4600 2913 3614
4 0 0 0 0 0 0 0 0 0
5 147 166 169 215 115 101 174 127 151
46886rh 46887rh 46888rh 46889rh 46890rh 46891rh 46892rh 46893rh 46895rh
1 16639 16072 0 16680 13367 0 25119 92 0
2 242543 258061 0 235530 196721 0 371037 1116 0
3 3294 3719 3899 4173 4008 3037 5967 3459 4262
4 0 0 0 0 0 0 0 0 0
5 139 128 210 127 156 116 260 161 189
46896rh 46897rh
1 15934 0
2 235343 6
3 3923 3486
4 0 0
5 179 117
As you can see, the gene ids are ENSEBL IDs. There is some risk that these may not be unique. Let’s check whether any of the gene symbols are duplicated. We will sum the number of duplicated gene symbols.
sum(duplicated(rownames(counts)))
[1] 0
The sum equals zero, so there are no duplicated gene symbols, which is good. Similarly, samples should be unique. Once again, let’s verify this:
sum(duplicated(colnames(counts)))
[1] 0
Formatting the count matrix
Now, as we see that gene_id is in first column of count matrix, but we will need only count data in matrix, so we will change the gene_id column to rownames.
Converting the gene_id as rownames of count matrix
counts <- counts %>% column_to_rownames(.,var="gene_id") %>% as.data.frame()
let’s confirm if change is done correctly
head(counts,n=5)
32043rh 32044rh 32046rh 32047rh 32048rh 32049rh 32050rh
ENSG00000080815 22554 0 0 0 16700 0 0
ENSG00000142192 344489 4 0 1 260935 6 8
ENSMUSG00000000001 5061 3483 3941 3088 2756 3067 2711
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 208 162 138 127 95 154 165
32052rh 32053rh 32057rh 32059rh 32061rh 32062rh 32065rh
ENSG00000080815 19748 14023 0 17062 0 15986 10
ENSG00000142192 337456 206851 1 264748 0 252248 172
ENSMUSG00000000001 3334 3841 4068 3306 4076 3732 3940
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 124 103 164 116 108 134 204
32067rh 32068rh 32070rh 32073rh 32074rh 32075rh 32078rh
ENSG00000080815 0 18584 1 0 0 22783 17029
ENSG00000142192 4 300398 4 2 9 342655 280968
ENSMUSG00000000001 4238 3257 3351 3449 4654 4844 3132
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 239 148 159 167 157 211 162
32081rh 32088rh 32640rh 46105rh 46106rh 46107rh 46108rh
ENSG00000080815 16626 15573 12721 4 0 0 0
ENSG00000142192 258597 243373 188818 19 0 0 1
ENSMUSG00000000001 3334 3639 3355 4191 3058 4265 3248
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 149 160 103 158 167 199 113
46109rh 46110rh 46111rh 46112rh 46113rh 46115rh 46121rh
ENSG00000080815 0 0 17931 0 19087 0 0
ENSG00000142192 5 1 293409 8 273704 1 0
ENSMUSG00000000001 3638 3747 3971 3192 3805 3753 4134
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 168 175 203 158 108 110 179
46131rh 46132rh 46134rh 46138rh 46141rh 46142rh 46862rh
ENSG00000080815 0 12703 18833 0 18702 17666 0
ENSG00000142192 1 187975 285048 0 284499 250600 0
ENSMUSG00000000001 3059 3116 3853 3682 2844 3466 3442
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 137 145 183 171 138 88 154
46863rh 46865rh 46866rh 46867rh 46868rh 46871rh 46872rh
ENSG00000080815 14834 10546 10830 10316 10638 15248 0
ENSG00000142192 218494 169516 152769 151732 190150 229063 6
ENSMUSG00000000001 3300 3242 3872 3656 3739 3473 3154
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 157 131 152 152 155 140 80
46873rh 46874rh 46875rh 46876rh 46877rh 46878rh 46879rh
ENSG00000080815 0 11608 11561 0 0 12683 15613
ENSG00000142192 1 165941 171303 0 2 183058 216122
ENSMUSG00000000001 5510 3657 4121 3422 3829 3996 4324
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 240 148 112 147 166 169 215
46881rh 46882rh 46883rh 46884rh 46885rh 46886rh 46887rh
ENSG00000080815 0 14084 20753 0 0 16639 16072
ENSG00000142192 0 199448 306081 0 5 242543 258061
ENSMUSG00000000001 2592 2606 4600 2913 3614 3294 3719
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 115 101 174 127 151 139 128
46888rh 46889rh 46890rh 46891rh 46892rh 46893rh 46895rh
ENSG00000080815 0 16680 13367 0 25119 92 0
ENSG00000142192 0 235530 196721 0 371037 1116 0
ENSMUSG00000000001 3899 4173 4008 3037 5967 3459 4262
ENSMUSG00000000003 0 0 0 0 0 0 0
ENSMUSG00000000028 210 127 156 116 260 161 189
46896rh 46897rh
ENSG00000080815 15934 0
ENSG00000142192 235343 6
ENSMUSG00000000001 3923 3486
ENSMUSG00000000003 0 0
ENSMUSG00000000028 179 117
As you can see from count table there are some genes that start with “ENSG” and others start with “ENSMUSG”. “ENSG” referes to human gene ENSEMBL id and “ENSMUSG” refer to mouse ENSEMBL id. Let’s check how many gene_ids are NOT from the mouse genome by searching for the string “MUS” (as in Mus musculus) in the rownames of count matrix
counts[,1:6] %>%
filter(!str_detect(rownames(.), "MUS"))
32043rh 32044rh 32046rh 32047rh 32048rh 32049rh
ENSG00000080815 22554 0 0 0 16700 0
ENSG00000142192 344489 4 0 1 260935 6
Ok, so we see there are two human genes in out count matrix. Why? What genes are they?
Briefly, 5xFAD mouse strain harbors two human transgenes APP (“ENSG00000142192”) and PSEN1 (“ENSG00000080815”) and inserted into exon 2 of the mouse Thy1 gene. To validate 5XFAD strain and capture expression of human transgene APP and PS1, a custom mouse genomic sequences was created and we quantified expression of human as well as mouse App (“ENSMUSG00000022892”) and Psen1 (“ENSMUSG00000019969”) genes by our MODEL-AD RNA-Seq pipeline.
Validation of 5xFAD mouse strain
#First we convert the dataframe to longer format and join our covariates by MouseID
count_tpose <- counts %>%
rownames_to_column(.,var="gene_id") %>%
filter(gene_id %in% c("ENSG00000080815","ENSMUSG00000019969","ENSG00000142192","ENSMUSG00000022892")) %>%
pivot_longer(.,cols = -"gene_id",names_to = "specimenID",values_to="counts") %>% as.data.frame() %>%
left_join(covars ,by="specimenID") %>% as.data.frame()
head(count_tpose)
gene_id specimenID counts individualID sex genotype
1 ENSG00000080815 32043rh 22554 32043 female 5XFAD_carrier
2 ENSG00000080815 32044rh 0 32044 male 5XFAD_noncarrier
3 ENSG00000080815 32046rh 0 32046 male 5XFAD_noncarrier
4 ENSG00000080815 32047rh 0 32047 male 5XFAD_noncarrier
5 ENSG00000080815 32048rh 16700 32048 female 5XFAD_carrier
6 ENSG00000080815 32049rh 0 32049 female 5XFAD_noncarrier
timepoint
1 12 mo
2 12 mo
3 12 mo
4 12 mo
5 12 mo
6 12 mo
#make the age column a factor and re-order the levels
count_tpose$timepoint <- factor(count_tpose$timepoint,levels=c("4 mo","6 mo","12 mo"))
# rename the gene id to gene symbol
count_tpose$gene_id[count_tpose$gene_id %in% "ENSG00000142192"] <- "Human APP"
count_tpose$gene_id[count_tpose$gene_id %in% "ENSG00000080815"] <- "Human PSEN1"
count_tpose$gene_id[count_tpose$gene_id %in% "ENSMUSG00000022892"] <- "Mouse App"
count_tpose$gene_id[count_tpose$gene_id %in% "ENSMUSG00000019969"] <- "Mouse Psen1"
#Create simple box plots showing normalized counts by genotype and time point, faceted by sex.
count_tpose %>%
ggplot(aes(x=timepoint, y=counts, color=genotype)) +
geom_boxplot() +
geom_point(position=position_jitterdodge()) +
facet_wrap(~sex+gene_id) +theme_bw()
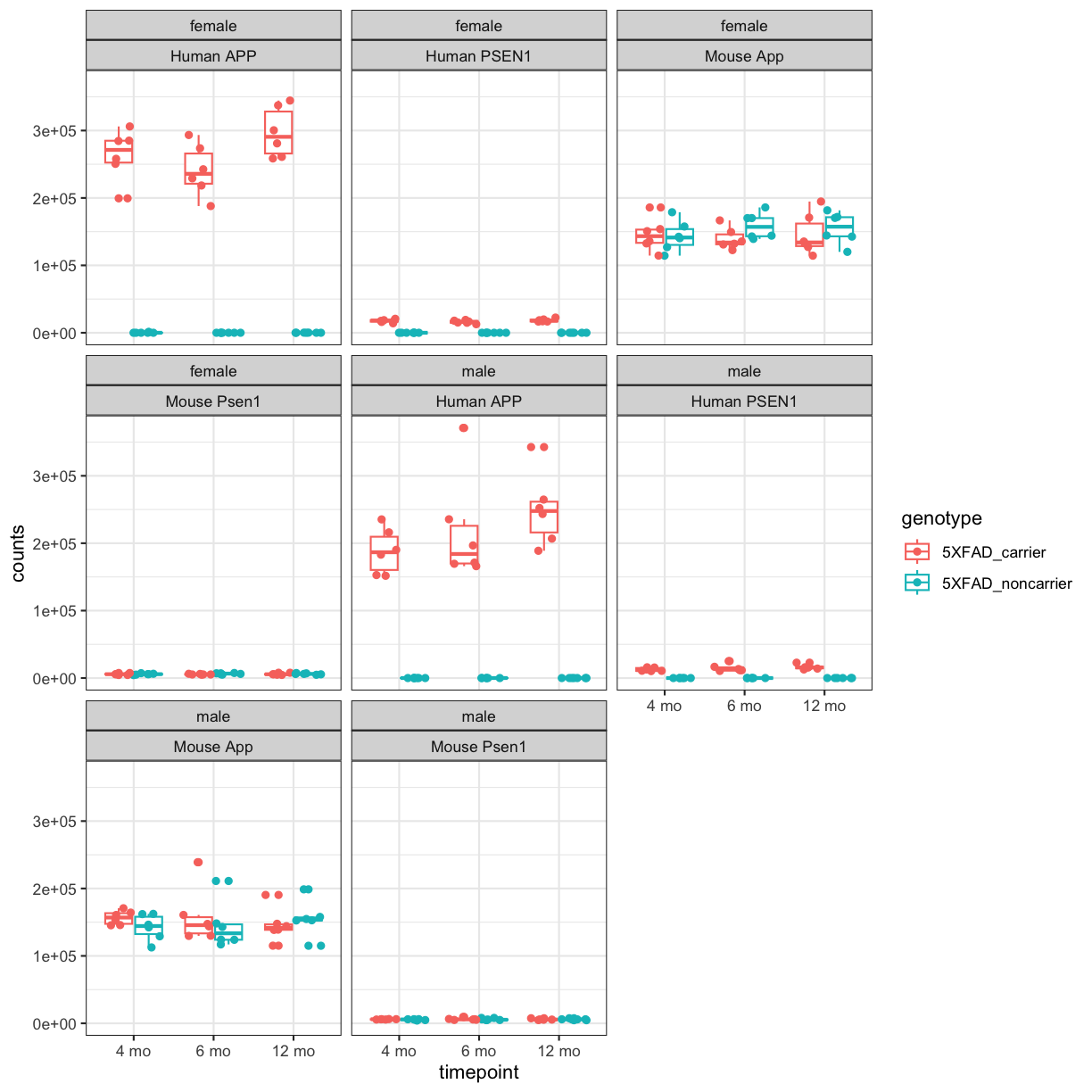
plot of chunk counts_boxplot
You will notice expression of Human APP is higher in 5XFAD carriers but lower in non-carriers. However mouse App expressed in both 5XFAD carrier and non-carrier.
We are going to sum the counts from both ortholgous genes (human APP and mouse App; human PSEN1 and mouse Psen1) and save summed expression as expression of mouse genes, respectively to match with gene names in control mice.
#merge mouse and human APP gene raw count
counts[rownames(counts) %in% "ENSMUSG00000022892",] <- counts[rownames(counts) %in% "ENSMUSG00000022892",] + counts[rownames(counts) %in% "ENSG00000142192",]
counts <- counts[!rownames(counts) %in% c("ENSG00000142192"),]
#merge mouse and human PS1 gene raw count
counts[rownames(counts) %in% "ENSMUSG00000019969",] <- counts[rownames(counts) %in% "ENSMUSG00000019969",] + counts[rownames(counts) %in% "ENSG00000080815",]
counts <- counts[!rownames(counts) %in% c("ENSG00000080815"),]
Let’s verify if expression of both human genes have been merged or not:
counts[,1:6] %>%
filter(!str_detect(rownames(.), "MUS"))
[1] 32043rh 32044rh 32046rh 32047rh 32048rh 32049rh
<0 rows> (or 0-length row.names)
What proportion of genes have zero counts in all samples?
gene_sums <- data.frame(gene_id = rownames(counts),
sums = Matrix::rowSums(counts))
sum(gene_sums$sums == 0)
[1] 9691
We can see that 9691 (17%) genes have no reads at all associated with them. In the next lesson, we will remove genes that have no counts in any samples.
Differential Analysis using DESeq2
Now, after exploring and formatting the data, We will look for differential expression between the control and 5xFAD mice at different ages for both sexes. The differentially expressed genes (DEGs) can inform our understanding of how the 5XFAD mutation affect the biological processes.
DESeq2 analysis consist of multiple steps. We are going to briefly understand some of the important steps using subset of data and then we will perform differential analysis on whole dataset. For detailed analysis see the DESeq2 tutorial.
First, order the data (so counts and metadata match) and save in another variable
rawdata <- counts[,sort(colnames(counts))]
metadata <- covars[sort(rownames(covars)),]
subset the counts matrix and sample metadata to include only 12 month old male mice. You can amend the code to compare wild type and 5XFAD mice from either sex, at any time point.
meta.12M.Male <- metadata[(metadata$sex=="male" & metadata$timepoint=='12 mo'),]
meta.12M.Male
individualID specimenID sex genotype timepoint
32044rh 32044 32044rh male 5XFAD_noncarrier 12 mo
32046rh 32046 32046rh male 5XFAD_noncarrier 12 mo
32047rh 32047 32047rh male 5XFAD_noncarrier 12 mo
32053rh 32053 32053rh male 5XFAD_carrier 12 mo
32059rh 32059 32059rh male 5XFAD_carrier 12 mo
32061rh 32061 32061rh male 5XFAD_noncarrier 12 mo
32062rh 32062 32062rh male 5XFAD_carrier 12 mo
32073rh 32073 32073rh male 5XFAD_noncarrier 12 mo
32074rh 32074 32074rh male 5XFAD_noncarrier 12 mo
32075rh 32075 32075rh male 5XFAD_carrier 12 mo
32088rh 32088 32088rh male 5XFAD_carrier 12 mo
32640rh 32640 32640rh male 5XFAD_carrier 12 mo
dat <- as.matrix(rawdata[,colnames(rawdata) %in% rownames(meta.12M.Male)])
colnames(dat)
[1] "32044rh" "32046rh" "32047rh" "32053rh" "32059rh" "32061rh" "32062rh"
[8] "32073rh" "32074rh" "32075rh" "32088rh" "32640rh"
rownames(meta.12M.Male)
[1] "32044rh" "32046rh" "32047rh" "32053rh" "32059rh" "32061rh" "32062rh"
[8] "32073rh" "32074rh" "32075rh" "32088rh" "32640rh"
match(colnames(dat),rownames(meta.12M.Male))
[1] 1 2 3 4 5 6 7 8 9 10 11 12
Next, we build the DESeqDataSet using the following function:
ddsHTSeq <- DESeqDataSetFromMatrix(countData=dat,
colData=meta.12M.Male,
design = ~genotype)
Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
design formula are characters, converting to factors
ddsHTSeq
class: DESeqDataSet
dim: 55487 12
metadata(1): version
assays(1): counts
rownames(55487): ENSMUSG00000000001 ENSMUSG00000000003 ...
ENSMUSG00000118487 ENSMUSG00000118488
rowData names(0):
colnames(12): 32044rh 32046rh ... 32088rh 32640rh
colData names(5): individualID specimenID sex genotype timepoint
Pre-filtering
While it is not necessary to pre-filter low count genes before running the DESeq2 functions, there are two reasons which make pre-filtering useful: by removing rows in which there are very few reads, we reduce the memory size of the dds data object, and we increase the speed of the transformation and testing functions within DESeq2. It can also improve visualizations, as features with no information for differential expression are not plotted.
Here we perform a minimal pre-filtering to keep only rows that have at least 10 reads total.
ddsHTSeq <- ddsHTSeq[rowSums(counts(ddsHTSeq)) >= 10,]
ddsHTSeq
class: DESeqDataSet
dim: 33059 12
metadata(1): version
assays(1): counts
rownames(33059): ENSMUSG00000000001 ENSMUSG00000000028 ...
ENSMUSG00000118486 ENSMUSG00000118487
rowData names(0):
colnames(12): 32044rh 32046rh ... 32088rh 32640rh
colData names(5): individualID specimenID sex genotype timepoint
Reference level
By default, R will choose a reference level for factors based on alphabetical order. Then, if you never tell the DESeq2 functions which level you want to compare against (e.g. which level represents the control group), the comparisons will be based on the alphabetical order of the levels.
specifying the reference-level to 5XFAD_noncarrier:
ddsHTSeq$genotype <- relevel(ddsHTSeq$genotype,ref="5XFAD_noncarrier")
Run the standard differential expression analysis steps that is wrapped into a single function, DESeq.
dds <- DESeq(ddsHTSeq,parallel = TRUE)
Results tables are generated using the function results, which extracts a results table with log2 fold changes, p values and adjusted p values. By default the argument alpha is set to 0.1. If the adjusted p value cutoff will be a value other than 0.1, alpha should be set to that value:
res <- results(dds,alpha=0.05) # setting 0.05 as significant threshold
res
log2 fold change (MLE): genotype 5XFAD carrier vs 5XFAD noncarrier
Wald test p-value: genotype 5XFAD carrier vs 5XFAD noncarrier
DataFrame with 33059 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSMUSG00000000001 3737.9009 0.0148125 0.0466948 0.317219 0.7510777
ENSMUSG00000000028 138.5635 -0.0712500 0.1550130 -0.459639 0.6457754
ENSMUSG00000000031 29.2983 0.6705922 0.3563441 1.881867 0.0598541
ENSMUSG00000000037 123.6482 -0.2184054 0.1554362 -1.405113 0.1599876
ENSMUSG00000000049 15.1733 0.3657555 0.3924376 0.932009 0.3513317
... ... ... ... ... ...
ENSMUSG00000118473 1.18647 -0.377971 1.531586 -0.246784 0.805075
ENSMUSG00000118477 59.10359 -0.144081 0.226690 -0.635586 0.525046
ENSMUSG00000118479 24.64566 -0.181992 0.341445 -0.533006 0.594029
ENSMUSG00000118486 1.92048 0.199838 1.253875 0.159376 0.873372
ENSMUSG00000118487 65.78311 -0.191362 0.218593 -0.875427 0.381342
padj
<numeric>
ENSMUSG00000000001 0.943421
ENSMUSG00000000028 0.913991
ENSMUSG00000000031 0.352346
ENSMUSG00000000037 0.566360
ENSMUSG00000000049 0.765640
... ...
ENSMUSG00000118473 NA
ENSMUSG00000118477 0.863565
ENSMUSG00000118479 0.893356
ENSMUSG00000118486 NA
ENSMUSG00000118487 0.785845
We can order our results table by the smallest p value:
resOrdered <- res[order(res$pvalue),]
head(resOrdered,n=10)
log2 fold change (MLE): genotype 5XFAD carrier vs 5XFAD noncarrier
Wald test p-value: genotype 5XFAD carrier vs 5XFAD noncarrier
DataFrame with 10 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSMUSG00000019969 13860.942 1.90740 0.0432685 44.0828 0.00000e+00
ENSMUSG00000030579 2367.096 2.61215 0.0749325 34.8600 2.99877e-266
ENSMUSG00000046805 7073.296 2.12247 0.0635035 33.4229 6.38260e-245
ENSMUSG00000032011 80423.476 1.36195 0.0424006 32.1210 2.24185e-226
ENSMUSG00000022892 271265.838 1.36140 0.0434167 31.3567 7.88666e-216
ENSMUSG00000038642 10323.969 1.69717 0.0549488 30.8864 1.81838e-209
ENSMUSG00000023992 2333.227 2.62290 0.0882818 29.7105 5.61657e-194
ENSMUSG00000079293 761.313 5.12514 0.1738382 29.4822 4.86644e-191
ENSMUSG00000040552 617.149 2.22726 0.0781799 28.4889 1.60622e-178
ENSMUSG00000069516 2604.926 2.34471 0.0847390 27.6697 1.61585e-168
padj
<numeric>
ENSMUSG00000019969 0.00000e+00
ENSMUSG00000030579 3.60828e-262
ENSMUSG00000046805 5.11991e-241
ENSMUSG00000032011 1.34876e-222
ENSMUSG00000022892 3.79585e-212
ENSMUSG00000038642 7.29321e-206
ENSMUSG00000023992 1.93090e-190
ENSMUSG00000079293 1.46389e-187
ENSMUSG00000040552 4.29486e-175
ENSMUSG00000069516 3.88853e-165
we can summarize some basic tallies using the summary function.
summary(res)
out of 33059 with nonzero total read count
adjusted p-value < 0.05
LFC > 0 (up) : 1098, 3.3%
LFC < 0 (down) : 505, 1.5%
outliers [1] : 33, 0.1%
low counts [2] : 8961, 27%
(mean count < 8)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
How many adjusted p-values were less than 0.05?
sum(res$padj < 0.05, na.rm=TRUE)
[1] 1603
Challenge 2
How many adjusted p-values were less than 0.1?
Solution to Challenge 2
sum(res$padj < 0.1, na.rm=TRUE)
Function to convert ensembleIDs to common gene names
We’ll use a package to translate mouse ENSEMBL IDS to gene names. Run this function and they will be called up when assembling results from the differential expression analysis
map_function.df <- function(x, inputtype, outputtype) {
mapIds(
org.Mm.eg.db,
keys = row.names(x),
column = outputtype,
keytype = inputtype,
multiVals = "first"
)
}
Generating Result table
All_res <- as.data.frame(res) %>%
mutate(symbol = map_function.df(res,"ENSEMBL","SYMBOL")) %>% ##run map_function to add symbol of gene corresponding to ENSEBL ID
mutate(EntrezGene = map_function.df(res,"ENSEMBL","ENTREZID")) %>% ##run map_function to add Entrez ID of gene corresponding to ENSEBL ID
dplyr::select("symbol", "EntrezGene","baseMean", "log2FoldChange", "lfcSE", "stat", "pvalue", "padj")
head(All_res)
symbol EntrezGene baseMean log2FoldChange lfcSE
ENSMUSG00000000001 Gnai3 14679 3737.90089 0.01481247 0.04669482
ENSMUSG00000000028 Cdc45 12544 138.56354 -0.07125004 0.15501305
ENSMUSG00000000031 H19 14955 29.29832 0.67059217 0.35634414
ENSMUSG00000000037 Scml2 107815 123.64823 -0.21840544 0.15543617
ENSMUSG00000000049 Apoh 11818 15.17325 0.36575554 0.39243763
ENSMUSG00000000056 Narf 67608 5017.30216 -0.06713961 0.04466809
stat pvalue padj
ENSMUSG00000000001 0.3172187 0.75107767 0.9434210
ENSMUSG00000000028 -0.4596390 0.64577536 0.9139907
ENSMUSG00000000031 1.8818667 0.05985412 0.3523457
ENSMUSG00000000037 -1.4051134 0.15998757 0.5663600
ENSMUSG00000000049 0.9320093 0.35133170 0.7656399
ENSMUSG00000000056 -1.5030778 0.13281898 0.5203330
Extracting genes that are significantly expressed
dseq_res <- subset(All_res[order(All_res$padj), ], padj < 0.05)
Wow! We have a lot of genes with apparently very strong statistically significant differences between the control and 5xFAD carrier.
dim(dseq_res)
[1] 1603 8
head(dseq_res)
symbol EntrezGene baseMean log2FoldChange lfcSE
ENSMUSG00000019969 Psen1 19164 13860.942 1.907397 0.04326854
ENSMUSG00000030579 Tyrobp 22177 2367.096 2.612152 0.07493255
ENSMUSG00000046805 Mpeg1 17476 7073.296 2.122467 0.06350346
ENSMUSG00000032011 Thy1 21838 80423.476 1.361953 0.04240065
ENSMUSG00000022892 App 11820 271265.838 1.361405 0.04341673
ENSMUSG00000038642 Ctss 13040 10323.969 1.697172 0.05494882
stat pvalue padj
ENSMUSG00000019969 44.08278 0.000000e+00 0.000000e+00
ENSMUSG00000030579 34.86005 2.998775e-266 3.608276e-262
ENSMUSG00000046805 33.42286 6.382596e-245 5.119906e-241
ENSMUSG00000032011 32.12104 2.241854e-226 1.348756e-222
ENSMUSG00000022892 31.35669 7.886662e-216 3.795850e-212
ENSMUSG00000038642 30.88641 1.818378e-209 7.293211e-206
Exploring and exporting results
Exporting results to CSV files
we can save results file into a csv file like this:
write.csv(All_res,file="../results/All_5xFAD_12months_male.csv")
write.csv(dseq_res,file="../results/DEG_5xFAD_12months_male.csv")
Volcano plot
We can visualize the differential expression results using Volcano plot function from EnhancedVolcano package. For the most basic volcano plot, only a single data-frame, data-matrix, or tibble of test results is required, containing point labels, log2FC, and adjusted or unadjusted P values. The default cut-off for log2FC is >|2|; the default cut-off for P value is 10e-6.
EnhancedVolcano(All_res,
lab = (All_res$symbol),
x = 'log2FoldChange',
y = 'padj',legendPosition = 'none',
title = 'Volcano plot:Differential Expression Results',
subtitle = '',
FCcutoff = 0.1,
pCutoff = 0.05,
xlim = c(-3, 6))
Warning: One or more p-values is 0. Converting to 10^-1 * current lowest
non-zero p-value...
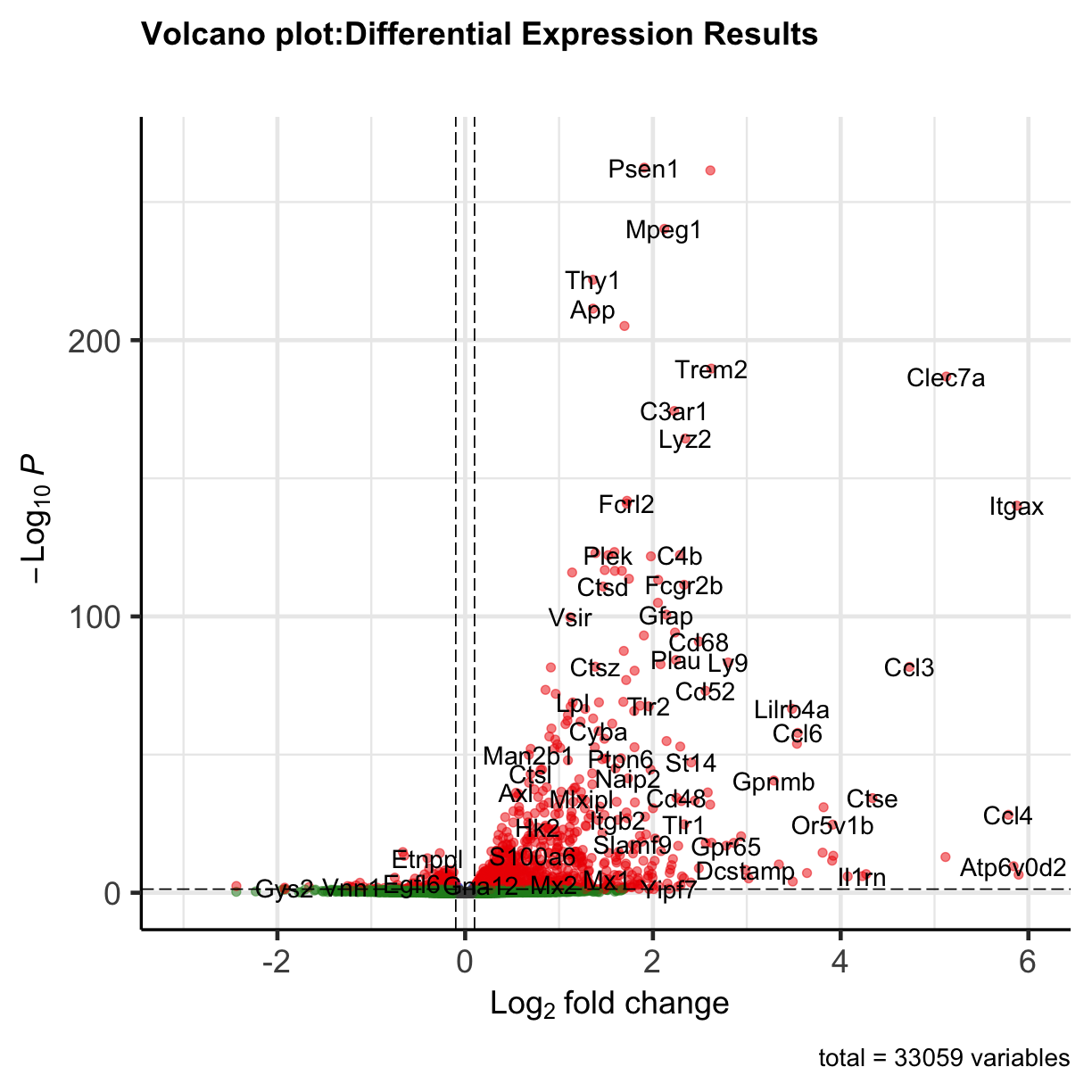
plot of chunk volcanoplot
You can see that some top significantly expressed are immune/inflammation-related genes such as Ctsd, C4b, Csf1 etc. These genes are upregulated in the 5XFAD strain.
Principal component plot of the samples
ddsHTSeq <- DESeqDataSetFromMatrix(countData=as.matrix(rawdata), colData=metadata, design= ~ genotype)
Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
design formula are characters, converting to factors
ddsHTSeq <- ddsHTSeq[rowSums(counts(ddsHTSeq)>1) >= 10, ]
dds <- DESeq(ddsHTSeq,parallel = TRUE)
estimating size factors
estimating dispersions
gene-wise dispersion estimates: 8 workers
mean-dispersion relationship
final dispersion estimates, fitting model and testing: 8 workers
-- replacing outliers and refitting for 42 genes
-- DESeq argument 'minReplicatesForReplace' = 7
-- original counts are preserved in counts(dds)
estimating dispersions
fitting model and testing
vsd <- varianceStabilizingTransformation(dds, blind=FALSE)
plotPCA(vsd, intgroup=c("genotype", "sex","timepoint"))
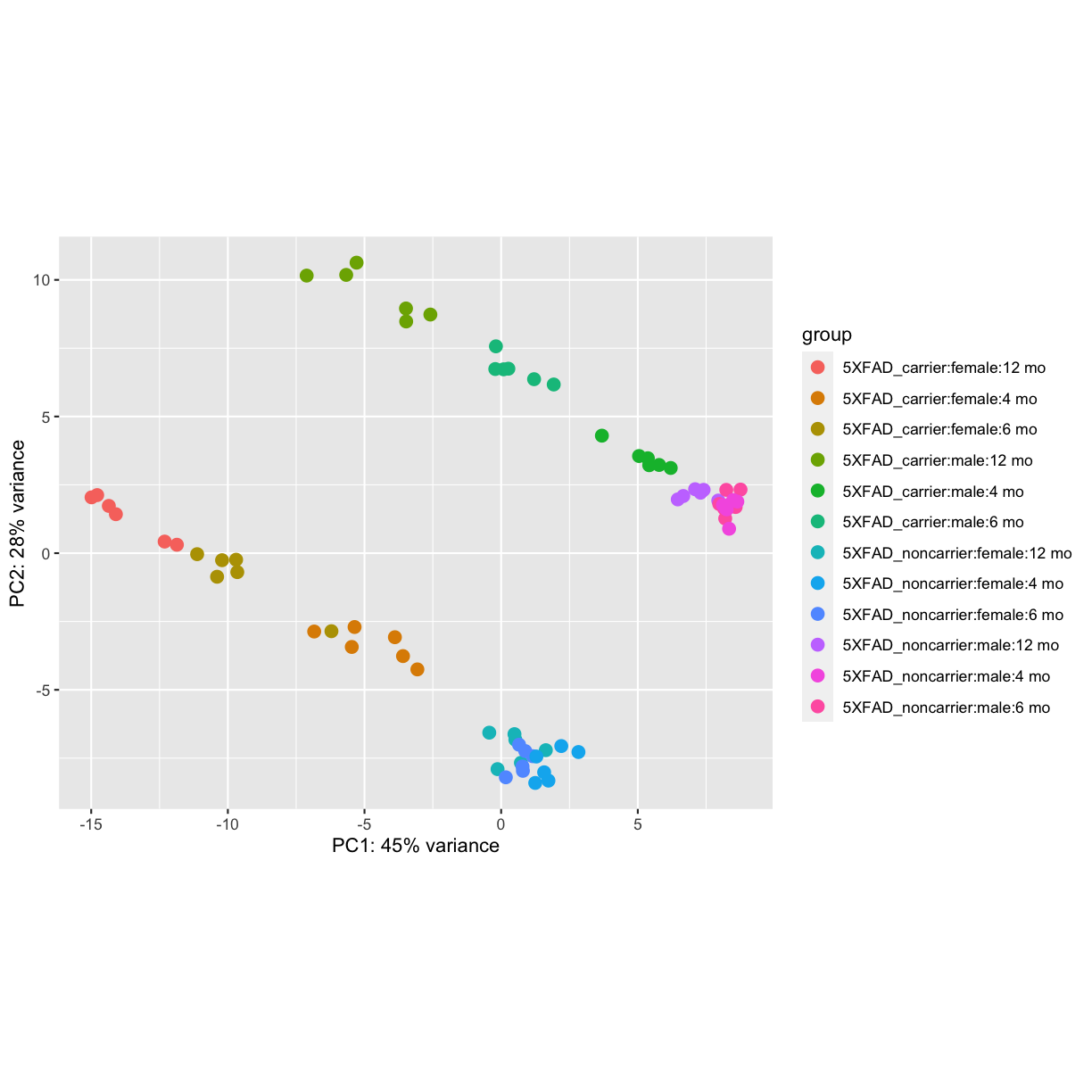
plot of chunk PCA
It is also possible to customize the PCA plot using the ggplot function.
pcaData <- plotPCA(vsd, intgroup=c("genotype", "sex","timepoint"), returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2,color=genotype, shape=sex)) +
geom_point(size=3) +
geom_text(aes(label=timepoint),hjust=0.5, vjust=2,size =3.5) +
labs(x= paste0("PC1: ",percentVar[1],"% variance"), y= paste0("PC2: ",percentVar[2],"% variance"))
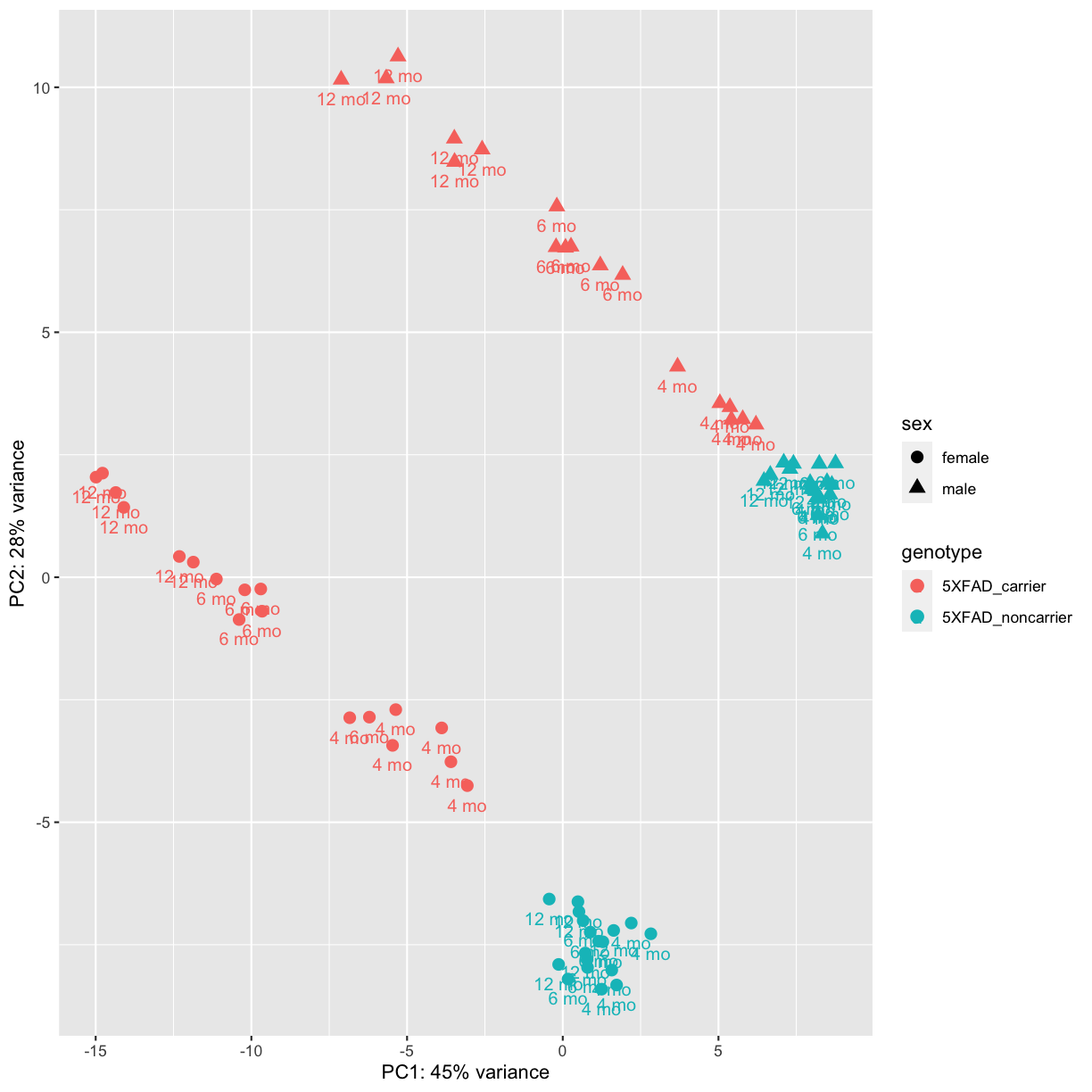
plot of chunk PCA2
PCA identified genotype and sex being a major source of variation in between 5XFAD and WT mice. Female and male samples clustered distinctly at all ages, suggesting the presence of sex-biased molecular changes in animals.
Function for Differential analysis using DESeq2
Finally, we can built a function for differential analysis that consist of all above discussed steps. It will require to input sorted raw count matrix, sample metadata and define the reference group.
DEG <- function(rawdata,meta,
include.batch = FALSE,
ref = ref) {
dseq_res <- data.frame()
All_res <- data.frame()
if (include.batch) {
cat("Including batch as covariate\n")
design_formula <- ~ Batch + genotype
}
else{
design_formula <- ~ genotype
}
dat2 <- as.matrix(rawdata[, colnames(rawdata) %in% rownames(meta)])
ddsHTSeq <-
DESeqDataSetFromMatrix(countData = dat2,
colData = meta,
design = design_formula)
ddsHTSeq <- ddsHTSeq[rowSums(counts(ddsHTSeq)) >= 10, ]
ddsHTSeq$genotype <- relevel(ddsHTSeq$genotype, ref = ref)
dds <- DESeq(ddsHTSeq, parallel = TRUE)
res <- results(dds, alpha = 0.05)
#summary(res)
res$symbol <- map_function.df(res,"ENSEMBL","SYMBOL")
res$EntrezGene <- map_function.df(res,"ENSEMBL","ENTREZID")
All_res <<- as.data.frame(res[, c("symbol", "EntrezGene","baseMean", "log2FoldChange", "lfcSE", "stat", "pvalue", "padj")])
}
Let’s use this function to analyze all groups present in our data.
Differential Analysis of all groups
First, we add a Group column to our metadata table that will combine all variable of interest for that sample.
metadata$Group <- paste0(metadata$genotype,"-",metadata$sex,"-",metadata$timepoint)
unique(metadata$Group)
[1] "5XFAD_carrier-female-12 mo" "5XFAD_noncarrier-male-12 mo"
[3] "5XFAD_noncarrier-female-12 mo" "5XFAD_carrier-male-12 mo"
[5] "5XFAD_noncarrier-female-6 mo" "5XFAD_noncarrier-male-6 mo"
[7] "5XFAD_carrier-female-6 mo" "5XFAD_noncarrier-female-4 mo"
[9] "5XFAD_carrier-female-4 mo" "5XFAD_carrier-male-6 mo"
[11] "5XFAD_carrier-male-4 mo" "5XFAD_noncarrier-male-4 mo"
Next, we create a comparison table that has all cases and controls that we would like to compare with each other. Here I have made comparison group for age and sex-matched 5xFAD carriers vs 5xFAD_noncarriers:
comparisons <- data.frame(control=c("5XFAD_noncarrier-male-4 mo", "5XFAD_noncarrier-female-4 mo", "5XFAD_noncarrier-male-6 mo",
"5XFAD_noncarrier-female-6 mo","5XFAD_noncarrier-male-12 mo", "5XFAD_noncarrier-female-12 mo"),
case=c("5XFAD_carrier-male-4 mo", "5XFAD_carrier-female-4 mo", "5XFAD_carrier-male-6 mo",
"5XFAD_carrier-female-6 mo","5XFAD_carrier-male-12 mo", "5XFAD_carrier-female-12 mo")
)
comparisons
control case
1 5XFAD_noncarrier-male-4 mo 5XFAD_carrier-male-4 mo
2 5XFAD_noncarrier-female-4 mo 5XFAD_carrier-female-4 mo
3 5XFAD_noncarrier-male-6 mo 5XFAD_carrier-male-6 mo
4 5XFAD_noncarrier-female-6 mo 5XFAD_carrier-female-6 mo
5 5XFAD_noncarrier-male-12 mo 5XFAD_carrier-male-12 mo
6 5XFAD_noncarrier-female-12 mo 5XFAD_carrier-female-12 mo
Finally, we implement our DEG function on each comparison and store the result table in a list and data frame:
# initiate an empty list and data frame to save results
DE_5xFAD.list <- list()
DE_5xFAD.df <- data.frame()
for (i in 1:nrow(comparisons))
{
meta <- metadata[metadata$Group %in% comparisons[i,],]
DEG(rawdata,meta,ref = "5XFAD_noncarrier")
#append results in data frame
DE_5xFAD.df <- rbind(DE_5xFAD.df,All_res %>% mutate(model="5xFAD",sex=unique(meta$sex),age=unique(meta$timepoint)))
#append results in list
DE_5xFAD.list[[i]] <- All_res
names(DE_5xFAD.list)[i] <- paste0(comparisons[i,2])
}
Let’s explore the result stored in our list:
names(DE_5xFAD.list)
[1] "5XFAD_carrier-male-4 mo" "5XFAD_carrier-female-4 mo"
[3] "5XFAD_carrier-male-6 mo" "5XFAD_carrier-female-6 mo"
[5] "5XFAD_carrier-male-12 mo" "5XFAD_carrier-female-12 mo"
We can easily extract result table for any group of interest by using $ and name of group. Let’s check top few rows from 5XFAD_carrier-male-4 mo group:
head(DE_5xFAD.list$`5XFAD_carrier-male-4 mo`)
symbol EntrezGene baseMean log2FoldChange lfcSE
ENSMUSG00000000001 Gnai3 14679 3707.53159 -0.023085865 0.0381646
ENSMUSG00000000028 Cdc45 12544 159.76225 -0.009444949 0.1322609
ENSMUSG00000000031 H19 14955 35.96987 0.453401555 0.2785245
ENSMUSG00000000037 Scml2 107815 126.82414 0.089394561 0.1377406
ENSMUSG00000000049 Apoh 11818 19.99721 0.115325837 0.3154875
ENSMUSG00000000056 Narf 67608 5344.21741 -0.100413295 0.0381182
stat pvalue padj
ENSMUSG00000000001 -0.60490264 0.545243691 0.9999518
ENSMUSG00000000028 -0.07141148 0.943070275 0.9999518
ENSMUSG00000000031 1.62786960 0.103552539 0.9999518
ENSMUSG00000000037 0.64900659 0.516334116 0.9999518
ENSMUSG00000000049 0.36554806 0.714702337 0.9999518
ENSMUSG00000000056 -2.63426085 0.008432068 0.5696800
Let’s check the result stored as dataframe:
head(DE_5xFAD.df)
symbol EntrezGene baseMean log2FoldChange lfcSE
ENSMUSG00000000001 Gnai3 14679 3707.53159 -0.023085865 0.0381646
ENSMUSG00000000028 Cdc45 12544 159.76225 -0.009444949 0.1322609
ENSMUSG00000000031 H19 14955 35.96987 0.453401555 0.2785245
ENSMUSG00000000037 Scml2 107815 126.82414 0.089394561 0.1377406
ENSMUSG00000000049 Apoh 11818 19.99721 0.115325837 0.3154875
ENSMUSG00000000056 Narf 67608 5344.21741 -0.100413295 0.0381182
stat pvalue padj model sex age
ENSMUSG00000000001 -0.60490264 0.545243691 0.9999518 5xFAD male 4 mo
ENSMUSG00000000028 -0.07141148 0.943070275 0.9999518 5xFAD male 4 mo
ENSMUSG00000000031 1.62786960 0.103552539 0.9999518 5xFAD male 4 mo
ENSMUSG00000000037 0.64900659 0.516334116 0.9999518 5xFAD male 4 mo
ENSMUSG00000000049 0.36554806 0.714702337 0.9999518 5xFAD male 4 mo
ENSMUSG00000000056 -2.63426085 0.008432068 0.5696800 5xFAD male 4 mo
Check if result is present for all ages:
unique((DE_5xFAD.df$age))
[1] "4 mo" "6 mo" "12 mo"
Check if result is present for both sexes:
unique((DE_5xFAD.df$sex))
[1] "male" "female"
Check number of genes in each group:
count(DE_5xFAD.df,model,sex,age)
model sex age n
1 5xFAD female 12 mo 33120
2 5xFAD female 4 mo 32930
3 5xFAD female 6 mo 33249
4 5xFAD male 12 mo 33059
5 5xFAD male 4 mo 33119
6 5xFAD male 6 mo 33375
Check number of genes significantly differentially expressed in all cases compared to age and sex-matched controls:
degs.up <- map(DE_5xFAD.list, ~length(which(.x$padj<0.05 & .x$log2FoldChange>0)))
degs.down <- map(DE_5xFAD.list, ~length(which(.x$padj<0.05 & .x$log2FoldChange<0)))
deg <- data.frame(Cases=names(degs.up), Up_DEGs.pval.05=as.vector(unlist(degs.up)),Down_DEGs.pval.05=as.vector(unlist(degs.down)))
knitr::kable(deg)
| Cases | Up_DEGs.pval.05 | Down_DEGs.pval.05 |
|---|---|---|
| 5XFAD_carrier-male-4 mo | 86 | 11 |
| 5XFAD_carrier-female-4 mo | 522 | 90 |
| 5XFAD_carrier-male-6 mo | 714 | 488 |
| 5XFAD_carrier-female-6 mo | 1081 | 409 |
| 5XFAD_carrier-male-12 mo | 1098 | 505 |
| 5XFAD_carrier-female-12 mo | 1494 | 1023 |
Interestingly, in females more genes are differentially expressed at 4 months and with age more genes are differentially expressed. Male mice is catching up female mice at later time-point.
Pathway Enrichment
We may wish to look for enrichment of biological pathways in a list of differentially expressed genes. Here we will test for enrichment of KEGG pathways using using enrichKEGG function in clusterProfiler package.
dat <- list(FAD_M_4=subset(DE_5xFAD.list$`5XFAD_carrier-male-4 mo`[order(DE_5xFAD.list$`5XFAD_carrier-male-4 mo`$padj), ], padj < 0.05) %>% pull(EntrezGene),
FAD_F_4=subset(DE_5xFAD.list$`5XFAD_carrier-female-4 mo`[order(DE_5xFAD.list$`5XFAD_carrier-female-4 mo`$padj), ], padj < 0.05) %>% pull(EntrezGene))
## perform enrichment analysis
enrich_pathway <- compareCluster(dat,
fun = "enrichKEGG",
pvalueCutoff = 0.05,
organism = "mmu"
)
enrich_pathway@compareClusterResult$Description <- gsub(" - Mus musculus \\(house mouse)","",enrich_pathway@compareClusterResult$Description)
Let’s plot top enriched functions using dotplot function of clusterProfiler package.
clusterProfiler::dotplot(enrich_pathway,showCategory=10,font.size = 14,title="Enriched KEGG Pathways")
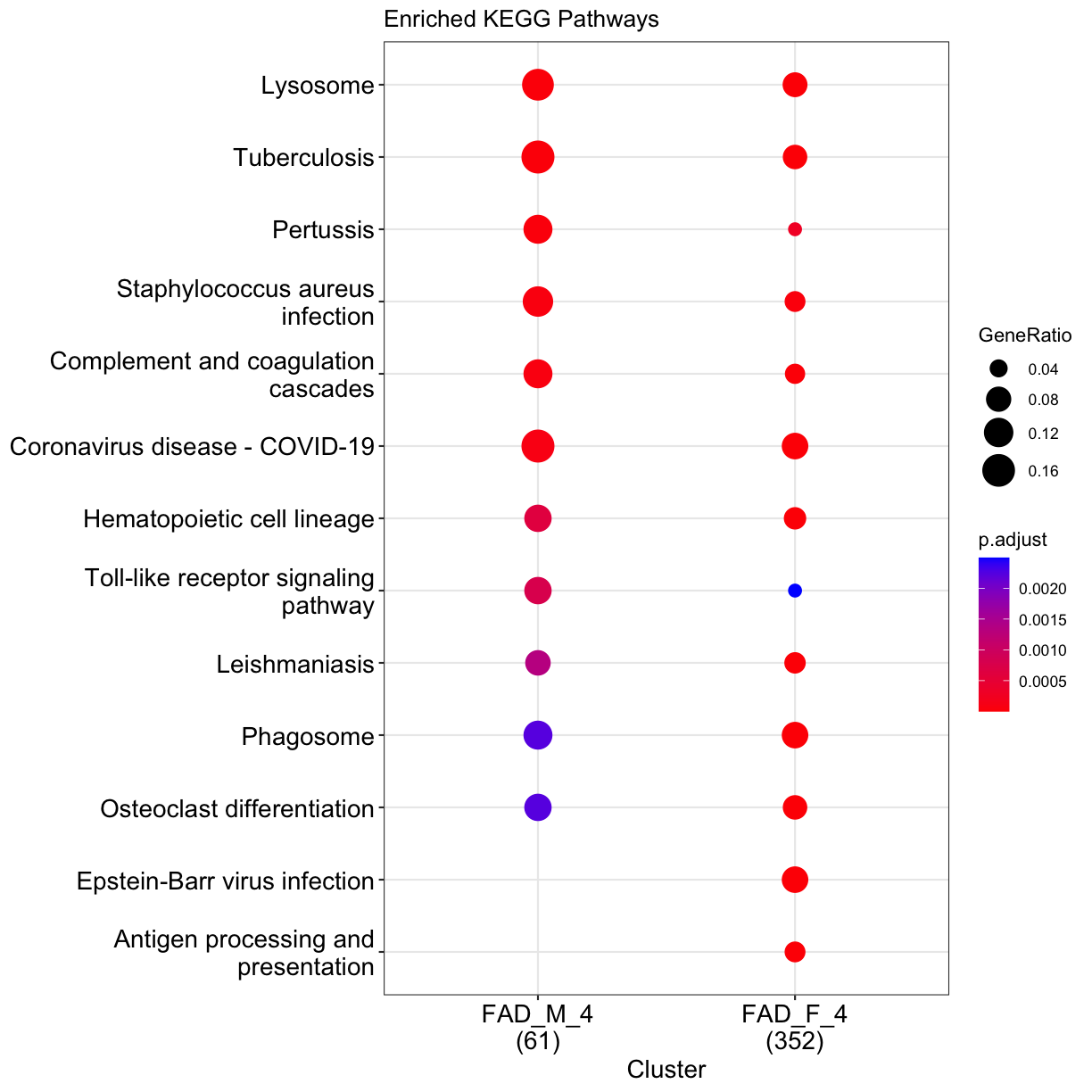
plot of chunk pathways
What does this plot infer?
Save Data for Next Lesson
We will use the results data in the next lesson. Save it now and we will load it at the beginning of the next lesson. We will use R’s save command to save the objects in compressed, binary format. The save command is useful when you want to save multiple objects in one file.
save(DE_5xFAD.df,DE_5xFAD.list,file="../results/DEAnalysis_5XFAD.Rdata")
Challenge 3
Draw volcano plot for 6 months old female 5xFAD_carrier ?
Solution to Challenge 3
EnhancedVolcano(DE_5xFAD.list$`5XFAD_carrier-female-6 mo, lab = (DE_5xFAD.list$`5XFAD_carrier-female-6 mo`$symbol), x = 'log2FoldChange', y = 'padj',legendPosition = 'none', title = 'Volcano plot:Differential Expression Results', subtitle = '', FCcutoff = 0.1, pCutoff = 0.05, xlim = c(-5, 5))
Loading RNA-Seq data from LOAD1 (APOE4*Trem2 Mouse Model)
There is another RNA-Seq data from LOAD1 mouse cohort. This cohort has three distinct mice strains: *APOE4: humanized Apoe knock-in strain, mouse Apoe allele was replaced by human APOE gene sequence; *Trem2.R47H:knock out mutant of the Trem2, R47H point mutation with two silent mutation and *APOE4.Trem2.R47H (LOAD1): double mutant strain carries humanized APOE (isoform 4) knock in mutation and R47H point mutation of the Trem2 gene.
Let’s read the count data and sample metadata as we did for 5XFAD samples.
counts <- read.delim("../data/htseqcounts_APTR.txt", check.names = FALSE)
ind_meta <- read.csv("../data/metadata_RNASEQ_ALLJAX_402Samples.csv")
Let’s look at the first few rows.
head(ind_meta)
Mouse.ID Sex Genotype Age Batch
1 123 M C57BL/6J 4 1
2 181 F C57BL/6J 4 1
3 191 M C57BL/6J 4 1
4 193 M C57BL/6J 4 1
5 221 F C57BL/6J 4 1
6 225 F C57BL/6J 4 1
Let’s quickly explore the sample metadata:
dim(ind_meta)
[1] 402 5
How many strains are in this sample metadata?
table(ind_meta$Genotype)
5XFAD Apoe4 APOE4 APOE4Trem2 ApoeKO APP Bin1
36 6 57 59 6 26 6
C57BL/6J Cd2ap Clu Trem2
137 6 6 57
How many samples are in each genotype for each sex and age group?
ind_meta %>% group_by(Sex,Genotype,Age) %>% count()
# A tibble: 51 × 4
# Groups: Sex, Genotype, Age [51]
Sex Genotype Age n
<chr> <chr> <int> <int>
1 F 5XFAD 4 6
2 F 5XFAD 6 6
3 F 5XFAD 12 6
4 F APOE4 4 12
5 F APOE4 8 6
6 F APOE4 12 5
7 F APOE4 24 6
8 F APOE4Trem2 4 12
9 F APOE4Trem2 8 6
10 F APOE4Trem2 12 5
# ℹ 41 more rows
We notice that there is a column Batch in sample metadata.
Let’s see how many samples are in each batch.
table(ind_meta$Batch)
1 2 3 4 5 6
140 94 72 36 48 12
So, total 5 batches. In bulk RNA-Seq experiments, it is usually vital that we apply a correction for samples profiled in different batches. Due to tim-constraint we do not want to do it in this lesson. So, we will use samples from only one batch in this lesson.
Let’s check which genotype are in which batch?
table(ind_meta$Batch,ind_meta$Genotype)
5XFAD Apoe4 APOE4 APOE4Trem2 ApoeKO APP Bin1 C57BL/6J Cd2ap Clu Trem2
1 0 0 33 35 0 0 0 36 0 0 36
2 0 0 24 24 0 0 0 25 0 0 21
3 36 0 0 0 0 0 0 36 0 0 0
4 0 6 0 0 6 0 6 6 6 6 0
5 0 0 0 0 0 16 0 32 0 0 0
6 0 0 0 0 0 10 0 2 0 0 0
We are going to use samples from Batch 1 as it has more samples and contain most of our LOAD1 cohort samples that we are interested.
Subset sample metadata for batch 1 only and Convert Mouse ID column to rownames.
covar <- ind_meta %>% filter(Batch==1) %>% remove_rownames() %>% column_to_rownames(.,var="Mouse.ID")
How many rows and columns are there in covar?
dim(covar)
[1] 140 4
Let’s look at the first few rows.
head(covar)
Sex Genotype Age Batch
123 M C57BL/6J 4 1
181 F C57BL/6J 4 1
191 M C57BL/6J 4 1
193 M C57BL/6J 4 1
221 F C57BL/6J 4 1
225 F C57BL/6J 4 1
Check mouse strains and are there numbers in covar?
table(covar$Genotype)
APOE4 APOE4Trem2 C57BL/6J Trem2
33 35 36 36
Challenge 4
How many samples are in each genotype for each sex and age group?
Solution to Challenge 4
covar %>% group_by(Sex,Genotype,Age) %>% count()
Let’s explore count data.
head(counts,n=5)
gene_ID 116 123 12 181 18421 18422 18424 18425 18427 18428
1 ENSG00000130203 54 37 112 90 16 35 29 36 6 6
2 ENSMUSG00000000001 2347 2648 2302 2585 3291 2256 2791 3029 1880 2298
3 ENSMUSG00000000003 0 0 0 0 0 0 0 0 0 0
4 ENSMUSG00000000028 89 114 60 113 109 72 76 86 73 93
5 ENSMUSG00000000031 47 20 7 18 20 17 12 16 6 12
18429 18430 18431 18432 18465 18469 18472 191 193 19682 19696 19700 20246
1 6 8 21 10 75147 106325 14 47 82 21898 46442 45987 0
2 2677 1884 2023 1745 3554 4040 4438 2494 2996 2545 2736 2851 4255
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 73 88 56 67 123 181 106 107 102 76 96 117 186
5 12 4 19 13 63 18 36 15 67 14 24 41 33
20248 20322 20357 20440 20443 20465 20466 20467 20495 20801 20804 20817
1 0 34116 46491 75376 77544 52 52 30 25511 117185 108849 108754
2 3247 2185 3238 3482 3903 2282 3075 2597 2124 4801 3994 4299
3 0 0 0 0 0 0 0 0 0 0 0 0
4 121 72 84 106 194 111 88 95 60 197 167 163
5 32 22 14 51 30 29 34 11 22 44 32 36
20822 20837 20875 20877 20879 20882 20887 20892 20893 20894 20895 20898 20899
1 70324 78966 78796 74305 80772 94969 4 48 62 32 39 76 12
2 3236 4267 3871 2534 3601 3543 2405 2282 2279 2600 2553 2971 1984
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 201 131 162 118 156 96 64 88 124 122 94 76 57
5 19 15 43 16 13 43 17 17 6 8 13 44 11
22001 22005 22078 22079 22083 22085 22087 22088 22090 22094 22095 22096 22097
1 51479 40252 52870 25029 31474 37812 36563 26525 48147 31663 26470 45566 55277
2 3161 2867 3400 2356 3121 3433 2569 1983 2933 2303 2190 2876 3571
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 89 127 112 89 116 102 127 86 108 84 105 67 124
5 16 10 16 12 24 21 9 9 8 14 20 65 30
22098 22144 22149 22168 22171 22172 22174 221 22514 22523 225 22705 22711
1 34337 36095 42308 34796 41583 30591 47146 41 27832 43621 64 30363 26211
2 2205 2732 2336 2094 2309 2214 2863 2605 2384 2536 3133 2091 1859
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 94 110 70 56 127 104 121 103 89 114 117 108 48
5 10 16 4 9 20 16 39 16 38 36 17 9 12
22712 22723 22746 22754 22755 22770 22869 23110 23156 23161 23168 23170 23184
1 30339 53178 37198 34847 48921 30110 43811 83574 74727 62068 85174 63851 73676
2 2487 2574 2763 2607 2740 3087 3298 4001 3578 3430 3365 2862 3198
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 91 111 95 123 73 105 130 135 134 187 149 126 142
5 24 39 25 3 29 14 45 82 14 12 29 8 33
23409 23410 23412 23414 23415 23450 23451 23454 239 242 251 252 25
1 75553 59847 71130 67167 91016 5 11 14 234 35 43 133 47
2 3537 3043 3514 3189 3944 2472 2226 2884 4137 2394 2945 2800 1989
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 115 134 131 102 194 61 94 137 125 156 80 87 66
5 36 30 34 35 44 20 22 26 7 21 35 100 5
26305 26313 26318 26330 26332 26334 26339 26346 26347 26617 26623 26624 27104
1 0 0 0 1 7 0 0 0 6 36805 38549 42588 39
2 3491 4493 3175 3577 3536 4017 3868 3685 2881 2483 2513 2992 2459
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 132 213 136 149 141 162 121 129 110 69 90 110 88
5 34 69 35 29 21 28 54 43 9 32 13 45 32
27168 27192 27194 27208 27209 272 27354 27355 27356 27357 27385 27386 27392
1 47 33724 40868 55 26 74 85 79 49 61 53 124 43
2 2358 2153 3043 2383 2339 2933 3605 2466 2813 3246 2996 2508 2517
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 87 98 112 107 55 100 124 94 68 150 123 88 97
5 10 12 30 26 20 13 233 29 24 35 20 32 18
27394 27395 27396 27397 27398 27399 27488 27495 27499 27504 27507 27509 27510
1 77 69 37 40 18 52 42890 53940 40501 21691 27115 32449 18986
2 3436 3214 3143 1757 2670 2282 2992 3289 2878 2306 2665 2912 2181
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 146 87 101 52 94 103 92 121 67 79 78 109 56
5 28 13 31 3 10 27 24 42 28 4 14 28 11
27585 27586 27590 27593 27594 27596 27598 27599 27600 27707 27708 27 28129
1 34478 40459 32038 20594 46469 27813 28542 25804 35189 31781 28269 32 104
2 2372 3032 2757 1967 3634 2270 2784 2398 2986 2281 2777 2324 2679
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 59 98 118 97 122 55 93 65 114 106 97 54 138
5 23 48 22 6 39 5 8 5 28 36 32 10 706
288810708 289457705 289461928 289470196 289478142 289482201 289494346
1 35 0 55 23 36 58 0
2 2091 3096 2148 2507 2615 2514 4320
3 0 0 0 0 0 0 0
4 63 116 87 131 55 99 214
5 14 28 16 20 28 8 19
289535121 289576914 289666353 289674340 299 30269 30270 30271 30 344 347
1 64 65 83 10 26 62 26 49 76 41 6
2 2567 3353 2985 2079 2381 3073 2374 2823 4084 3185 3295
3 0 0 0 0 0 0 0 0 0 0 0
4 95 134 109 79 126 102 89 94 128 170 123
5 20 31 30 12 8 29 10 21 49 26 19
35 364 367 393 394 399 3 400 40407 40408 40416 40423 40425 40434
1 35251 20 7 58 19 17 27 10 63779 91374 19 1 0 0
2 2796 3799 3344 5148 3413 2649 2290 3987 3191 4044 3726 3041 2857 3724
3 0 0 0 0 0 0 0 0 0 0 0 0 0 0
4 122 173 186 205 156 103 121 179 100 186 180 161 103 172
5 12 36 32 71 24 17 11 30 17 54 54 45 37 36
40438 40439 40571 40573 40575 40576 40579 40580 40581 40585 40587 40590
1 0 0 128540 78818 55469 59 0 1 0 0 60212 58952
2 3685 2737 6846 4165 3458 2748 4149 3596 4379 3948 3197 3512
3 0 0 0 0 0 0 0 0 0 0 0 0
4 151 104 271 197 139 90 175 156 186 123 101 114
5 53 28 74 67 20 46 54 44 56 44 50 38
40591 40593 40595 40808 40823 40834 40837 40839 40845 40877 40880 42 44
1 78712 0 74833 69807 1 72482 84527 78285 77004 62765 94765 40950 22335
2 4050 3129 3799 3056 4161 3497 3606 3510 4246 3958 4091 2675 2471
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 169 120 139 75 175 148 138 160 146 139 201 131 86
5 68 33 35 19 45 29 39 21 21 62 37 13 34
46611 46618 46621 46623 46630 46642 46647 46648 46649 46650 46651 46652
1 61971 57335 66163 60289 0 68907 107855 0 0 0 0 0
2 3396 2746 2999 3303 3264 3407 4788 3566 3046 3240 4054 3681
3 0 0 0 0 0 0 0 0 0 0 0 0
4 158 149 149 120 111 167 193 140 160 97 135 172
5 20 26 56 29 34 11 41 22 37 6 53 51
46653 46654 46662 46686 46687 46688 46698 46 54 56 57 58 8
1 0 0 59014 0 9 1 58161 53318 41514 42211 37472 45864 77
2 3628 4260 2984 5382 3955 3028 2780 3244 2533 3570 2421 2677 3300
3 0 0 0 0 0 0 0 0 0 0 0 0 0
4 102 173 132 139 233 108 118 162 94 150 71 93 166
5 12 38 11 53 66 33 46 14 11 20 3 19 43
Converting the gene_id as rownames of count matrix
counts <- counts %>% column_to_rownames(.,var="gene_ID") %>% as.data.frame()
How many rows and columns are there in counts?
dim(counts)
[1] 55488 234
In the counts matrix, genes are in rows and samples are in columns.
There are 140 samples in batch 1 as we saw in covar and samples in counts matrix is 234. So, we need to subset counts matrix for samples in covar.
counts <- counts[,colnames(counts) %in% rownames(covar)]
dim(counts)
[1] 55488 140
Now there are count data from 140 samples present in metadata.
Check how many gene_ids are NOT from the mouse genome by searching for the string “MUS” (as in Mus musculus) in the rownames of count matrix
counts[,1:6] %>%
filter(!str_detect(rownames(.), "MUS"))
116 123 12 181 18421 18422
ENSG00000130203 54 37 112 90 16 35
We see entry of gene id “ENSG00000130203”. This is human APOE gene that has been quantified by our MODEL-AD RNA-Seq pipeline.
Here you can also check expression of mouse Apoe.
counts[rownames(counts) %in% "ENSMUSG00000002985",1:10]
116 123 12 181 18421 18422 18424 18425 18427 18428
ENSMUSG00000002985 44173 51114 45209 58427 72939 47959 37884 43401 49936 54232
Let’s visualize the expression of mouse and human APOE gene across all samples using ggplot2 package.
Validation of mouse strains
#First we convert the dataframe to longer format and join our covariates by MouseID
count_tpose <- counts %>%
rownames_to_column(.,var="gene_id") %>%
filter(gene_id %in% c("ENSG00000130203","ENSMUSG00000002985")) %>%
pivot_longer(.,cols = -"gene_id",names_to = "Mouse.ID",values_to="counts") %>% as.data.frame() %>%
left_join(ind_meta %>% mutate("Mouse.ID"=as.character(Mouse.ID)),by="Mouse.ID") %>% as.data.frame()
head(count_tpose)
gene_id Mouse.ID counts Sex Genotype Age Batch
1 ENSG00000130203 116 54 F Trem2 8 1
2 ENSG00000130203 123 37 M C57BL/6J 4 1
3 ENSG00000130203 12 112 M C57BL/6J 8 1
4 ENSG00000130203 181 90 F C57BL/6J 4 1
5 ENSG00000130203 18421 16 F Trem2 12 1
6 ENSG00000130203 18422 35 M Trem2 8 1
#make the age column a factor and re-order the levels
count_tpose$Age <- factor(count_tpose$Age,levels=c(4,8,12))
# rename the gene id to gene symbol
count_tpose$gene_id[count_tpose$gene_id %in% "ENSG00000130203"] <- "Human APOE"
count_tpose$gene_id[count_tpose$gene_id %in% "ENSMUSG00000002985"] <- "Mouse Apoe"
#Create simple box plots showing normalized counts by genotype and time point, faceted by sex.
count_tpose %>%
ggplot(aes(x=Age, y=counts, color=Genotype)) +
geom_boxplot() +
geom_point(position=position_jitterdodge()) +
facet_wrap(~Sex+gene_id) +theme_bw()
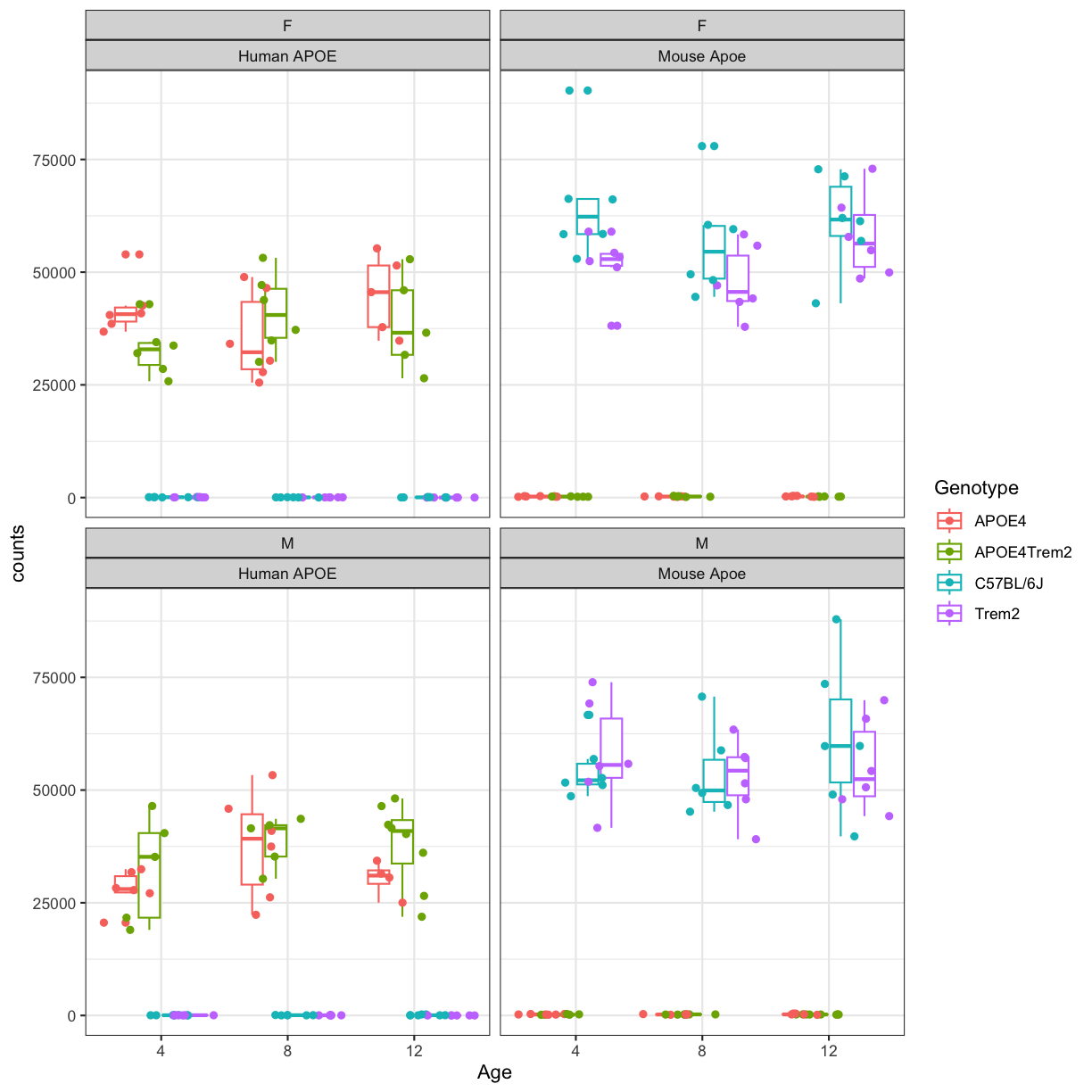
plot of chunk boxplot_APOE4_cohort
You will notice expression of mouse Apoe is higher in samples carrying humanized APOE gene sequences but lower in other samples and vice-versa for human APOE. This also suggest that mouse Apoe gene has been correctly replaced by human APOE gene sequences in APOE4 and APOE4Trem strains. This way we can validated the insertion of human transgene.
As before, we need to merge expression of human APOE and mouse Apoe:
counts[rownames(counts) %in% "ENSMUSG00000002985",] <- counts[rownames(counts) %in% "ENSMUSG00000002985",] + counts[rownames(counts) %in% "ENSG00000130203",]
counts <- counts[!rownames(counts) %in% c("ENSG00000130203"),]
Let’s verify
counts[,1:6] %>%
filter(!str_detect(rownames(.), "MUS"))
[1] 116 123 12 181 18421 18422
<0 rows> (or 0-length row.names)
Formatting the count and metadata for differential analysis
rawdata <- counts[,sort(colnames(counts))]
metadata <- covar[sort(rownames(covar)),]
## renaming column names
colnames(metadata) <- c("sex","genotype","timepoint","Batch")
metadata$sex[metadata$sex %in% "F"] <- "female"
metadata$sex[metadata$sex %in% "M"] <- "male"
Differential Analysis of all groups in LOAD1 cohort
Next, we create a comparison table that has all cases and controls that we would like to compare with each other. Here I have made comparison group for age and sex-matched distinct mouse strains vs C57BL/6J (control mice):
metadata$Group <- paste0(metadata$genotype,"-",metadata$sex,"-",metadata$timepoint)
comparisons <- data.frame(
control=rep(c("C57BL/6J-male-4", "C57BL/6J-female-4", "C57BL/6J-male-8", "C57BL/6J-female-8", "C57BL/6J-male-12", "C57BL/6J-female-12"),3),
case=c("APOE4-male-4", "APOE4-female-4", "APOE4-male-8", "APOE4-female-8", "APOE4-male-12", "APOE4-female-12",
"Trem2-male-4", "Trem2-female-4", "Trem2-male-8", "Trem2-female-8", "Trem2-male-12", "Trem2-female-12",
"APOE4Trem2-male-4", "APOE4Trem2-female-4", "APOE4Trem2-male-8", "APOE4Trem2-female-8", "APOE4Trem2-male-12", "APOE4Trem2-female-12")
)
head(comparisons)
control case
1 C57BL/6J-male-4 APOE4-male-4
2 C57BL/6J-female-4 APOE4-female-4
3 C57BL/6J-male-8 APOE4-male-8
4 C57BL/6J-female-8 APOE4-female-8
5 C57BL/6J-male-12 APOE4-male-12
6 C57BL/6J-female-12 APOE4-female-12
Finally, we implement our DEG function on each comparison and store the result table in a list and data frame:
DE_LOAD1.list <- list()
DE_LOAD1.df <- data.frame()
for (i in 1:nrow(comparisons)){
meta <- metadata[metadata$Group %in% comparisons[i,],]
DEG(rawdata,meta,ref = "C57BL/6J")
#append results in data frame
DE_LOAD1.df <- rbind(DE_LOAD1.df,All_res %>% mutate(model=gsub("-.*$","",comparisons[i,2])[1],sex=unique(meta$sex),age=unique(meta$timepoint)))
#append results in list
DE_LOAD1.list[[i]] <- All_res
names(DE_LOAD1.list)[i] <- paste0(comparisons[i,2])
}
save(DE_LOAD1.df,DE_LOAD1.list,file="../results/DEAnalysis_LOAD1.Rdata")
check number of genes significantly expressed (adjusted p =0.05) in all cases compared to age and sex-matched controls:
degs.up <- map(DE_LOAD1.list, ~length(which(.x$padj<0.05 & .x$log2FoldChange>0)))
degs.down <- map(DE_LOAD1.list, ~length(which(.x$padj<0.05 & .x$log2FoldChange<0)))
deg <- data.frame(comparison=names(degs.up), Up_DEGs.pval.05=as.vector(unlist(degs.up)),Down_DEGs.pval.05=as.vector(unlist(degs.down)))
knitr::kable(deg)
| comparison | Up_DEGs.pval.05 | Down_DEGs.pval.05 |
|---|---|---|
| APOE4-male-4 | 0 | 3 |
| APOE4-female-4 | 0 | 1 |
| APOE4-male-8 | 42 | 103 |
| APOE4-female-8 | 11 | 14 |
| APOE4-male-12 | 0 | 1 |
| APOE4-female-12 | 0 | 3 |
| Trem2-male-4 | 5 | 59 |
| Trem2-female-4 | 5 | 13 |
| Trem2-male-8 | 3 | 29 |
| Trem2-female-8 | 2 | 9 |
| Trem2-male-12 | 118 | 88 |
| Trem2-female-12 | 113 | 172 |
| APOE4Trem2-male-4 | 15 | 8 |
| APOE4Trem2-female-4 | 1 | 2 |
| APOE4Trem2-male-8 | 7 | 32 |
| APOE4Trem2-female-8 | 2 | 1 |
| APOE4Trem2-male-12 | 0 | 2 |
| APOE4Trem2-female-12 | 1 | 4 |
Challenge 5
Save the data for next lesson?
Solution to Challenge 5
save(DE_LOAD1.df,DE_LOAD1.list,file="../result/DEAnalysis_LOAD1.Rdata")
Session Info
sessionInfo()
R version 4.3.0 (2023-04-21)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Monterey 12.6.2
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] clusterProfiler_4.8.1 lubridate_1.9.2
[3] forcats_1.0.0 stringr_1.5.0
[5] dplyr_1.1.2 purrr_1.0.1
[7] readr_2.1.4 tidyr_1.3.0
[9] tibble_3.2.1 tidyverse_2.0.0
[11] EnhancedVolcano_1.18.0 ggrepel_0.9.3
[13] GO.db_3.17.0 org.Mm.eg.db_3.17.0
[15] AnnotationDbi_1.62.1 ggplot2_3.4.2
[17] DESeq2_1.40.1 SummarizedExperiment_1.30.2
[19] Biobase_2.60.0 MatrixGenerics_1.12.0
[21] matrixStats_1.0.0 GenomicRanges_1.52.0
[23] GenomeInfoDb_1.36.0 IRanges_2.34.0
[25] S4Vectors_0.38.1 BiocGenerics_0.46.0
[27] knitr_1.43
loaded via a namespace (and not attached):
[1] DBI_1.1.3 bitops_1.0-7 gson_0.1.0
[4] shadowtext_0.1.2 gridExtra_2.3 rlang_1.1.1
[7] magrittr_2.0.3 DOSE_3.26.1 compiler_4.3.0
[10] RSQLite_2.3.1 png_0.1-8 vctrs_0.6.2
[13] reshape2_1.4.4 pkgconfig_2.0.3 crayon_1.5.2
[16] fastmap_1.1.1 XVector_0.40.0 labeling_0.4.2
[19] ggraph_2.1.0 utf8_1.2.3 HDO.db_0.99.1
[22] tzdb_0.4.0 enrichplot_1.20.0 bit_4.0.5
[25] xfun_0.39 zlibbioc_1.46.0 cachem_1.0.8
[28] aplot_0.1.10 jsonlite_1.8.5 blob_1.2.4
[31] highr_0.10 DelayedArray_0.26.3 BiocParallel_1.34.2
[34] tweenr_2.0.2 parallel_4.3.0 R6_2.5.1
[37] RColorBrewer_1.1-3 stringi_1.7.12 GOSemSim_2.26.0
[40] Rcpp_1.0.10 downloader_0.4 Matrix_1.5-4
[43] splines_4.3.0 igraph_1.4.3 timechange_0.2.0
[46] tidyselect_1.2.0 viridis_0.6.3 qvalue_2.32.0
[49] codetools_0.2-19 lattice_0.21-8 plyr_1.8.8
[52] treeio_1.24.1 withr_2.5.0 KEGGREST_1.40.0
[55] evaluate_0.21 gridGraphics_0.5-1 scatterpie_0.2.1
[58] polyclip_1.10-4 Biostrings_2.68.1 ggtree_3.8.0
[61] pillar_1.9.0 ggfun_0.0.9 generics_0.1.3
[64] RCurl_1.98-1.12 hms_1.1.3 tidytree_0.4.2
[67] munsell_0.5.0 scales_1.2.1 glue_1.6.2
[70] lazyeval_0.2.2 tools_4.3.0 data.table_1.14.8
[73] fgsea_1.26.0 locfit_1.5-9.8 graphlayouts_1.0.0
[76] fastmatch_1.1-3 tidygraph_1.2.3 cowplot_1.1.1
[79] grid_4.3.0 ape_5.7-1 colorspace_2.1-0
[82] nlme_3.1-162 patchwork_1.1.2 GenomeInfoDbData_1.2.10
[85] ggforce_0.4.1 cli_3.6.1 fansi_1.0.4
[88] viridisLite_0.4.2 S4Arrays_1.0.4 gtable_0.3.3
[91] yulab.utils_0.0.6 digest_0.6.31 ggplotify_0.1.0
[94] farver_2.1.1 memoise_2.0.1 lifecycle_1.0.3
[97] httr_1.4.6 bit64_4.0.5 MASS_7.3-58.4
Key Points
Validate metadata prior to data analysis.
Use function for repeated differential expression analysis in multiple comparisons.